
 Tweet
Tweet
Hello,
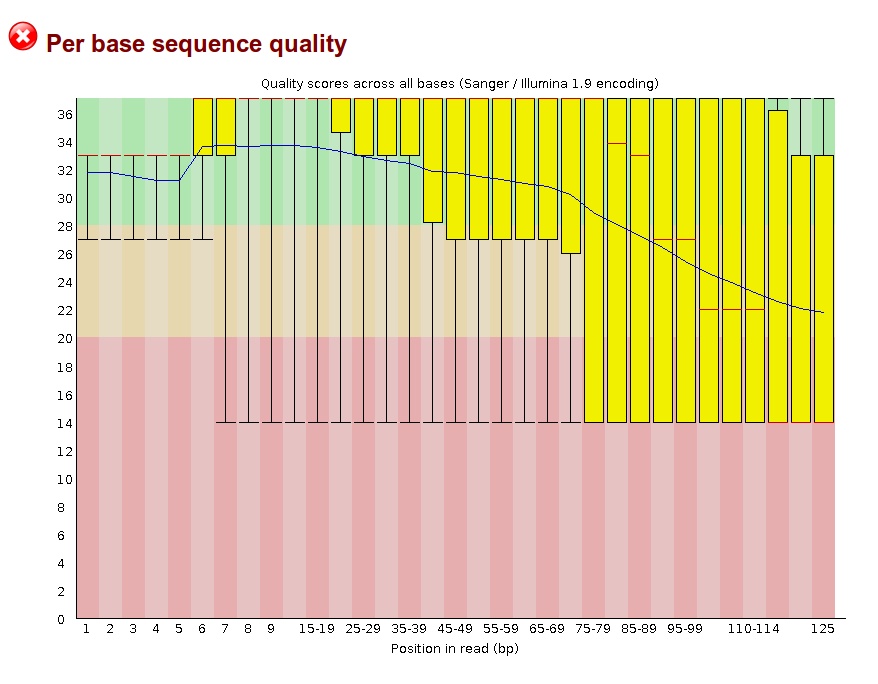
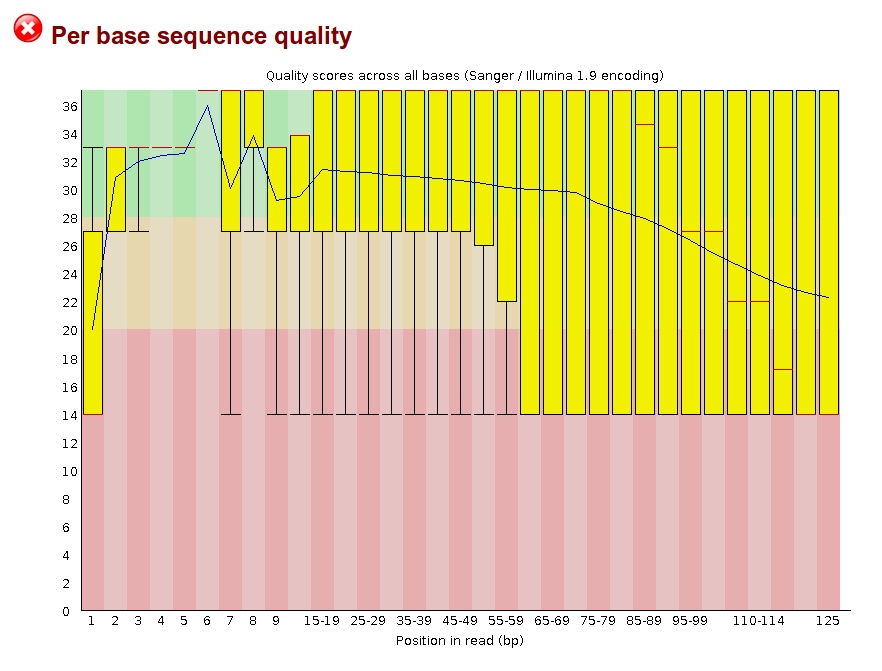
Thank you for taking the time to read my post. I currently have whole genome metagenomic data taken from the eye, with the reads being a mix of human DNA and a number of different bacterial species' DNA. With the reads being paired-end, I am wondering if it can be that two reads from a pair can be from the genome of two different organisms, thus making the paired-end alignment invalid with the reads being able to be aligned as single-ended? I tried doing alignment with Bowtie2, which has an option to look for single-ended alignments after paired-end alignments fail -- when I looked at the unique read IDs that were mapped, there was a fairly large increase (~30% or so), which is a good amount larger than I expected, based on past sequencing projects I've worked with.
When trimming the reads for quality, I am trying to decide the parameters for removing pairs where one read may have many bases removed, whether to keep these reads as single-ended, etc. Any help is greatly appreciated.
Thank you for taking the time to read my post. I currently have whole genome metagenomic data taken from the eye, with the reads being a mix of human DNA and a number of different bacterial species' DNA. With the reads being paired-end, I am wondering if it can be that two reads from a pair can be from the genome of two different organisms, thus making the paired-end alignment invalid with the reads being able to be aligned as single-ended? I tried doing alignment with Bowtie2, which has an option to look for single-ended alignments after paired-end alignments fail -- when I looked at the unique read IDs that were mapped, there was a fairly large increase (~30% or so), which is a good amount larger than I expected, based on past sequencing projects I've worked with.
When trimming the reads for quality, I am trying to decide the parameters for removing pairs where one read may have many bases removed, whether to keep these reads as single-ended, etc. Any help is greatly appreciated.




Comment