
 Tweet
Tweet
I'm trying to characterize the fragment length distribution of some Illumina data from the 1000 Genomes Project.
To do this, I downloaded the supplied BAM files (reads aligned with MAQ) for the mtDNA of the CEU trio daughter (to make sure rearrangements wouldn't be a problem.) Then I checked the distance between the two farthest ends of the mapped pairs.
I thought this would be fairly straightforward, but the mean and median aren't even close to the fragment size listed for some of the libraries. They're much lower than the listed fragment size. The distributions aren't even remotely normal, either.
For example, SRR001139 is supposed to have a fragment length of 676 with mate paired reads of size 47. The mean fragment length I estimate, however, is 234 with a standard deviation of 148.
Here is a histogram of the length distribution:
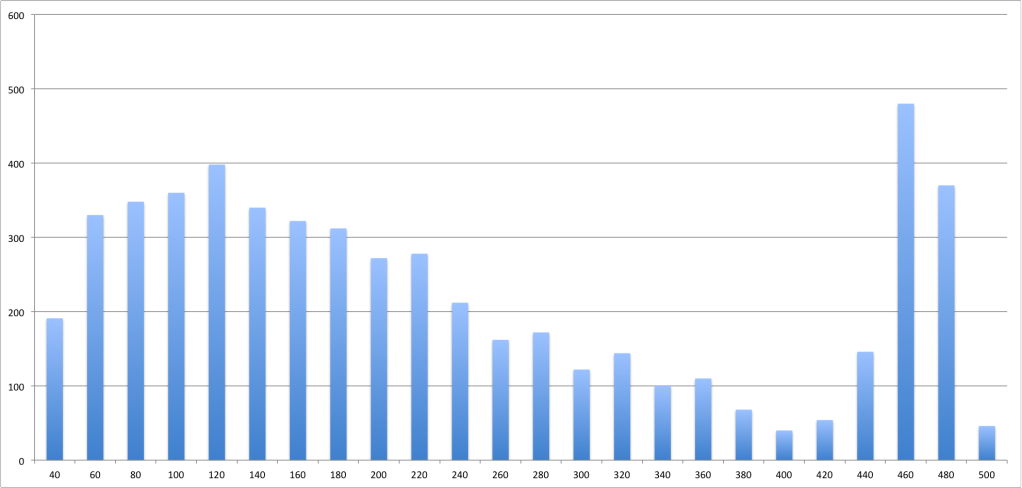
Is expected? Or am I making a mistake somewhere along the way in my reasoning? Does anyone know any references about the fragment lengths that result from Solexa sequencing?

Any help or suggestions would be much appreciated.
To do this, I downloaded the supplied BAM files (reads aligned with MAQ) for the mtDNA of the CEU trio daughter (to make sure rearrangements wouldn't be a problem.) Then I checked the distance between the two farthest ends of the mapped pairs.
I thought this would be fairly straightforward, but the mean and median aren't even close to the fragment size listed for some of the libraries. They're much lower than the listed fragment size. The distributions aren't even remotely normal, either.
For example, SRR001139 is supposed to have a fragment length of 676 with mate paired reads of size 47. The mean fragment length I estimate, however, is 234 with a standard deviation of 148.
Here is a histogram of the length distribution:
Is expected? Or am I making a mistake somewhere along the way in my reasoning? Does anyone know any references about the fragment lengths that result from Solexa sequencing?
Any help or suggestions would be much appreciated.


Comment