
 Tweet
Tweet
Greetings all!
Many thanks to everyone who has contributed to this very helpful and informative forum. This is my first post, so I figured I'd make things a bit interesting with a strange scenario I've recently encountered...
I'm currently undertaking library preparation using a bit of a hybrid of Illumina's TruSeq and MatePair kits and protocols. The goal is to generate three libraries of different insert sizes - roughly 300 bp, 3kb and 5kb. These different insert-size-libraries are then pooled at a specific ratio and run across multiple lanes on a HiSeq flow cell.
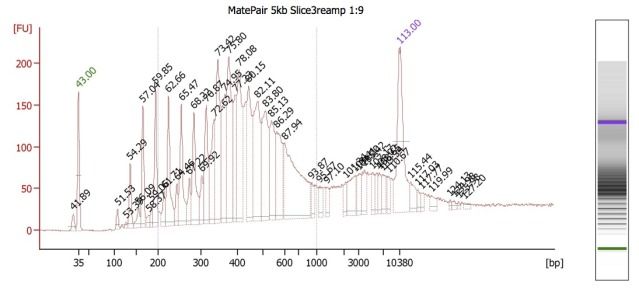
Because these different insert-size-libraries are pooled, they require indexed adapters to sort out their library (and thus, insert size) of origin. This is where the hybrid protocol comes in: the Mate Pair kit does not have indexed adapters, so we've integrated TruSeq adapters into this protocol. So far, this worked well for creating the 3kb libraries, but for some reason, this did NOT work well for the 5kb libraries. BioAnalyzer results (and agarose gels) showed the presence of many, regularly spaced, fragment bands.
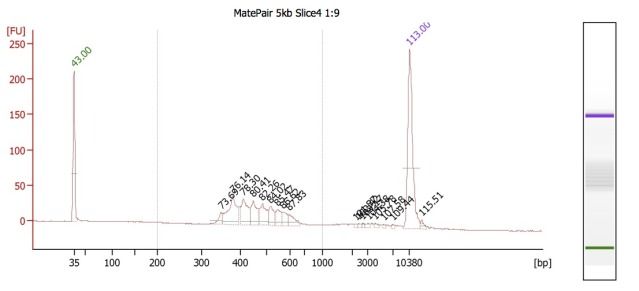
We believe that these are primer dimers/concatemers of some kind, but are puzzled on why they would appear for the 5kb libraries but not the 3kb ones. Both Mate Pair library types were set up at the same time, using the same DNA, and utilizing nearly identical workflow procedures.
Has anyone out there in Next-Generation-Sequencing-land experienced similar results??
Many thanks!
Paul
Many thanks to everyone who has contributed to this very helpful and informative forum. This is my first post, so I figured I'd make things a bit interesting with a strange scenario I've recently encountered...
I'm currently undertaking library preparation using a bit of a hybrid of Illumina's TruSeq and MatePair kits and protocols. The goal is to generate three libraries of different insert sizes - roughly 300 bp, 3kb and 5kb. These different insert-size-libraries are then pooled at a specific ratio and run across multiple lanes on a HiSeq flow cell.
Because these different insert-size-libraries are pooled, they require indexed adapters to sort out their library (and thus, insert size) of origin. This is where the hybrid protocol comes in: the Mate Pair kit does not have indexed adapters, so we've integrated TruSeq adapters into this protocol. So far, this worked well for creating the 3kb libraries, but for some reason, this did NOT work well for the 5kb libraries. BioAnalyzer results (and agarose gels) showed the presence of many, regularly spaced, fragment bands.
We believe that these are primer dimers/concatemers of some kind, but are puzzled on why they would appear for the 5kb libraries but not the 3kb ones. Both Mate Pair library types were set up at the same time, using the same DNA, and utilizing nearly identical workflow procedures.
Has anyone out there in Next-Generation-Sequencing-land experienced similar results??
Many thanks!
Paul





Comment