 Tweet
Tweet
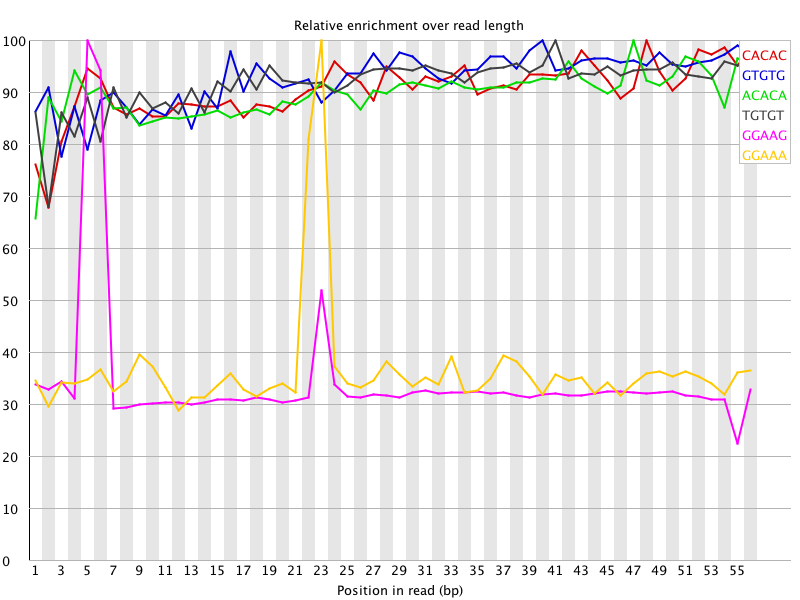
I ran FatsQC on a set of 4 Illumina PE exomes and got really weird graphs for the kmer distribution. I've never seen this pattern before - does anyone know what is going on and what to do about it?
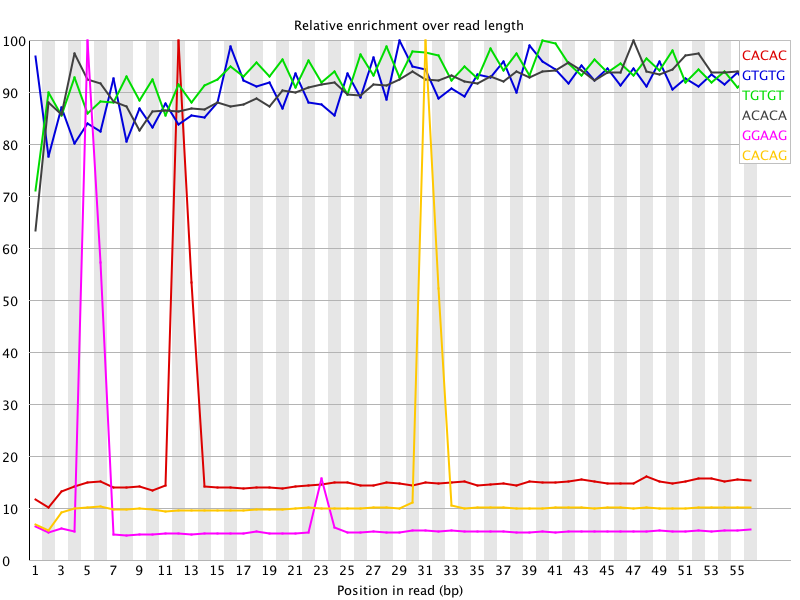
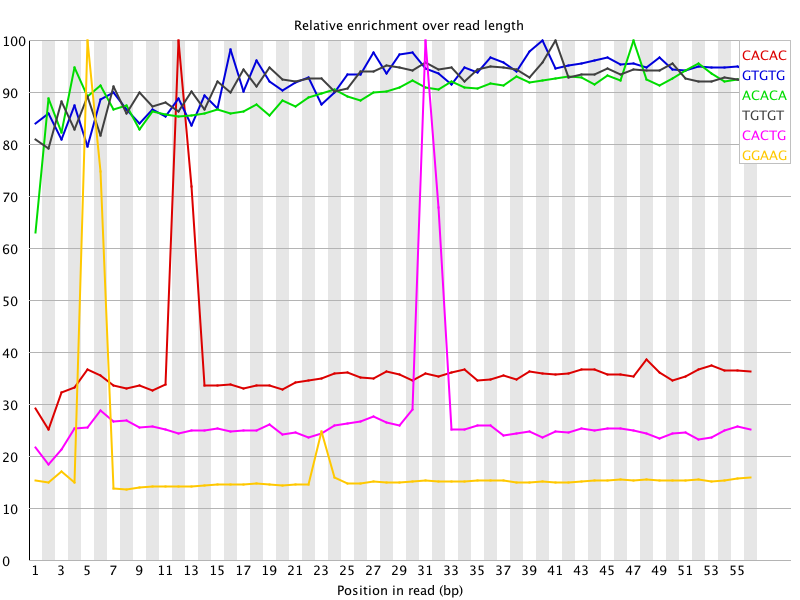
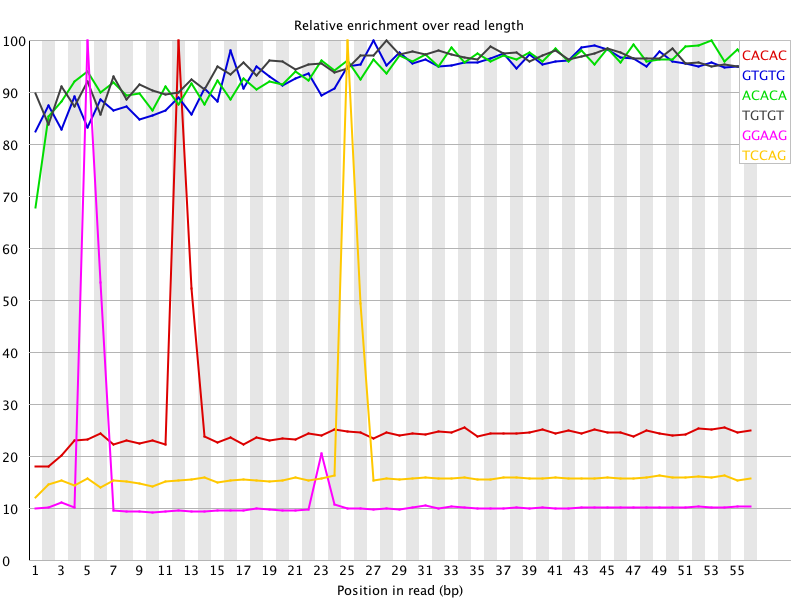
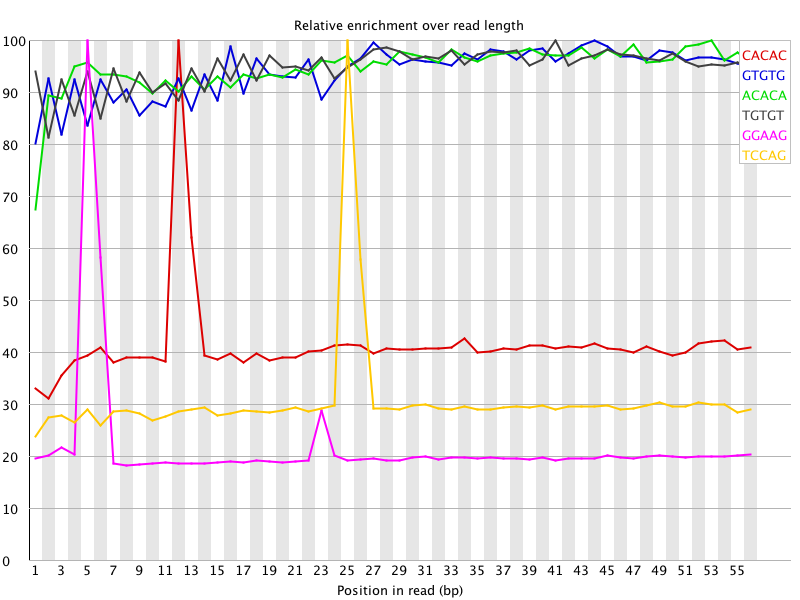
You are currently viewing the SEQanswers forums as a guest, which limits your access. Click here to register now, and join the discussion
Comment