 Tweet
Tweet
I was trying to use your MapView to look at some ELAND alignment results but didn't get very far. Since I'm new to all next generation sequencing, please excuse my ignorant questions:
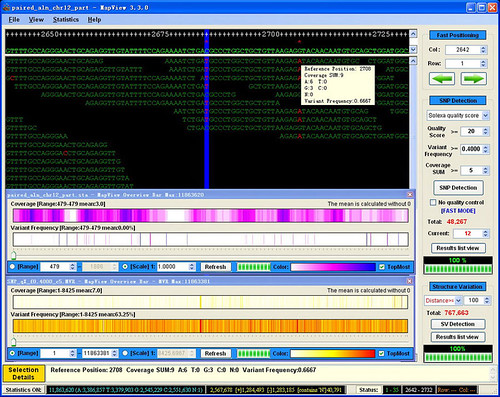
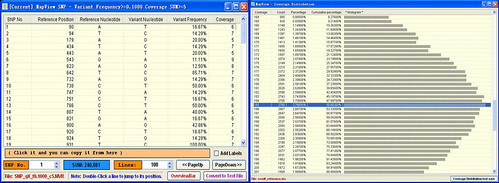
1. Which ELAND alignment file should I use in the MVFmaker? I tried both the s_*_*export.txt and s_*_*_sorted.txt together with my reference genome. All I got were red x and no short reads.
2. My Solexa data comes from a paired-end sequencing of a bacterial strain. How can I input the second read?
Thank you.
1. Which ELAND alignment file should I use in the MVFmaker? I tried both the s_*_*export.txt and s_*_*_sorted.txt together with my reference genome. All I got were red x and no short reads.
2. My Solexa data comes from a paired-end sequencing of a bacterial strain. How can I input the second read?
Thank you.

Comment