 Tweet
Tweet
Hi all,
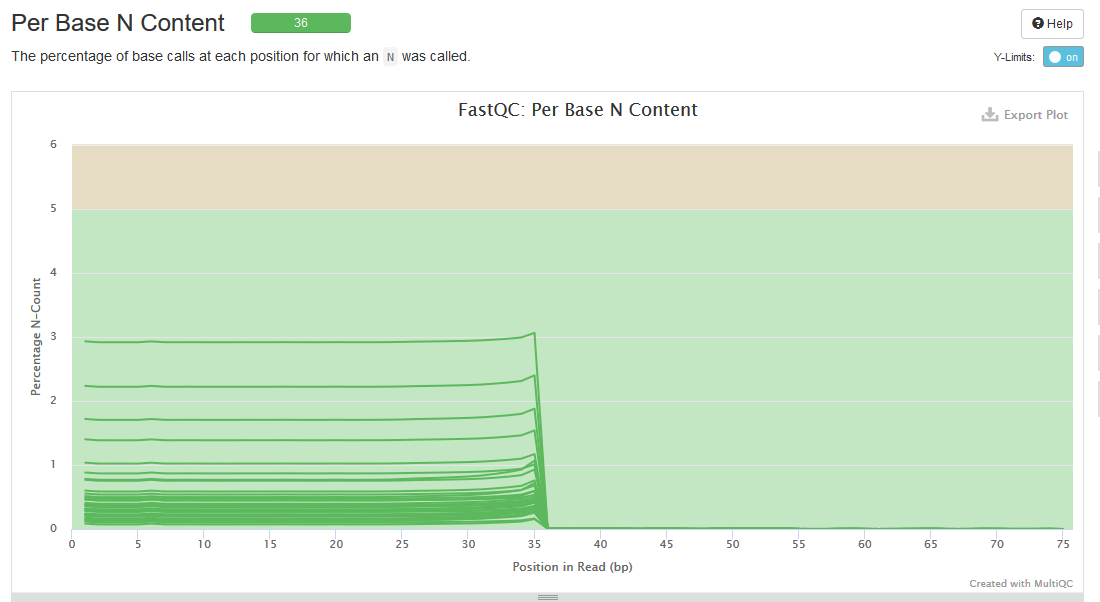
Just did some sequencing for a client and got a little odd plot using MultiQC for the "FastQ per base N-Content". It looks like the first 35 bp had problems with "N" then suddenly stopped. The percentage isn't high, but I don't understand what happened. Using trimmomatic removed these which appeared to be runs of "N". Anyone seen something like this before, or have an explanation for this?
Thanks,
-pete
Edit: We made these libraries using KAPA tagmentation kits, and there were no bad tiles (none) using FastQC.
Just did some sequencing for a client and got a little odd plot using MultiQC for the "FastQ per base N-Content". It looks like the first 35 bp had problems with "N" then suddenly stopped. The percentage isn't high, but I don't understand what happened. Using trimmomatic removed these which appeared to be runs of "N". Anyone seen something like this before, or have an explanation for this?

Thanks,
-pete
Edit: We made these libraries using KAPA tagmentation kits, and there were no bad tiles (none) using FastQC.


Comment