 Tweet
Tweet
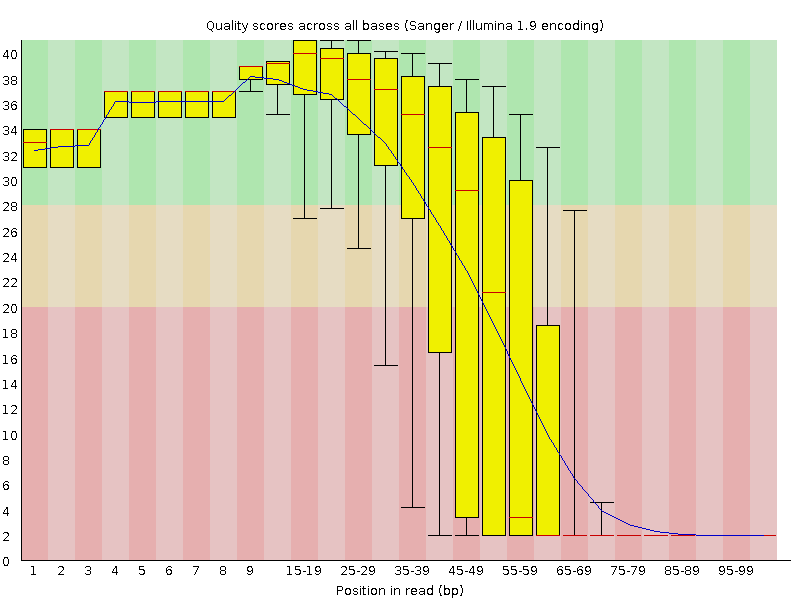
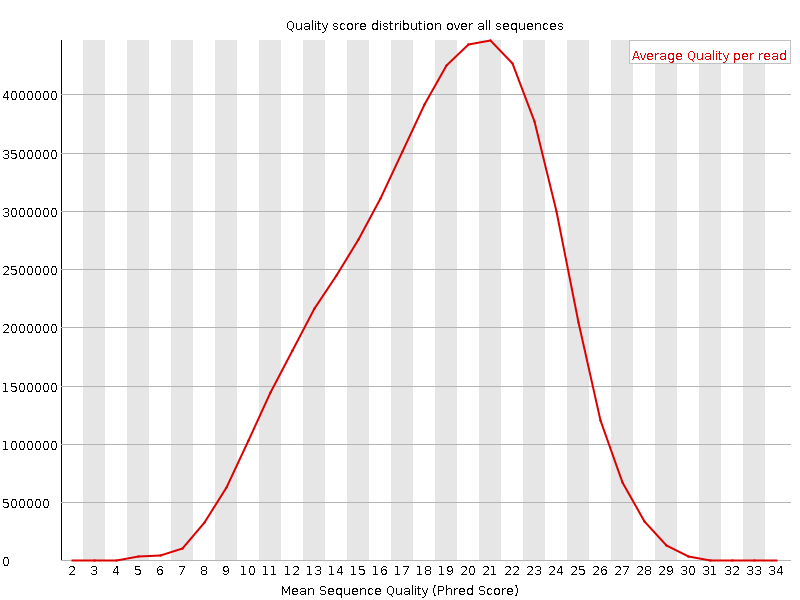
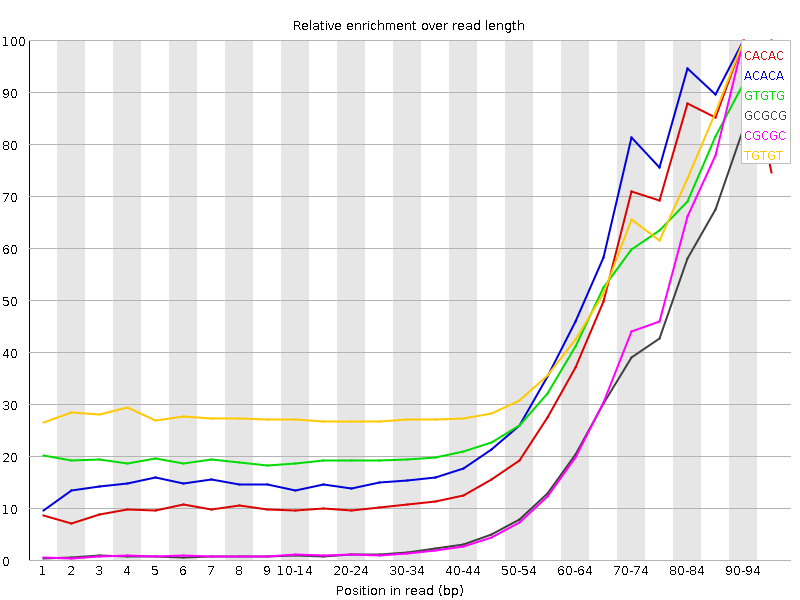
I have whole genome data in paired end fastq files. Before going for its alignment, I checked some of the samples for quality using FastQC but I'm getting strange results as seen in the attachments. Shall I move ahead with such data or need to trim these sequences or discard the data? I've majority of samples like this!
-
Thanks, -
Hi,
I would suggest you trim the leftmost part of the sequences. Given that you still have some high quality bases at the 3'-end, it will be good to set a Q-score threshold during trimming. By doing this you will end up with reads of varying lengths.
http://www.youtube.com/watch?v=bz93ReOv87Y
HTH -
Thanks, what about the sequences which ll be trimmed? Doesn't it has any impact on the results?Thanks,Comment
-
Which ever method you use should enable you retain the high quality 5'-ends of trimmed reads.Comment
-
I am not sure how much trimming will help you as your quality declines pretty rapidly and not very far into the reads. Try it, but I would also point this out to who ever did the sequencing (assuming it wasn't you).Comment
-
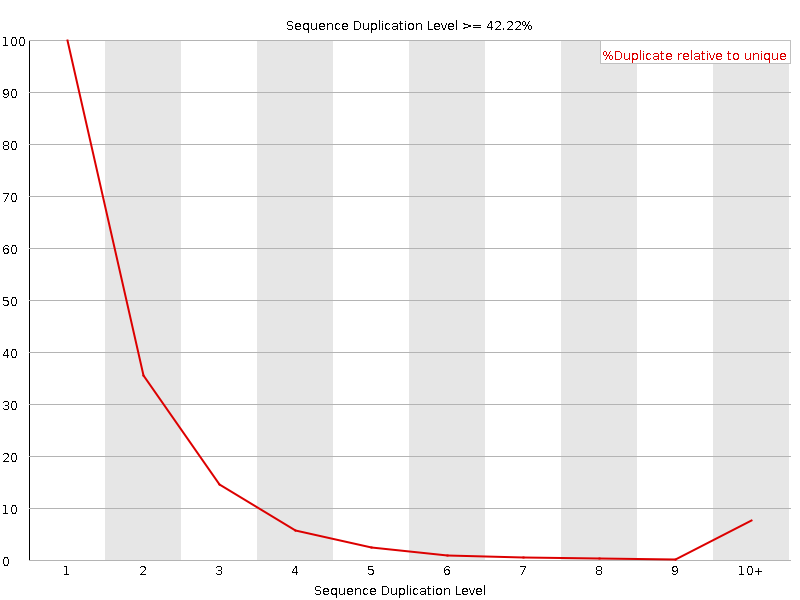
Your Kmer plot suggests that a reasonable proportion of your library contains very short inserts (we can't really tell what proportion from the graph - you'd need the accompanying table for that).
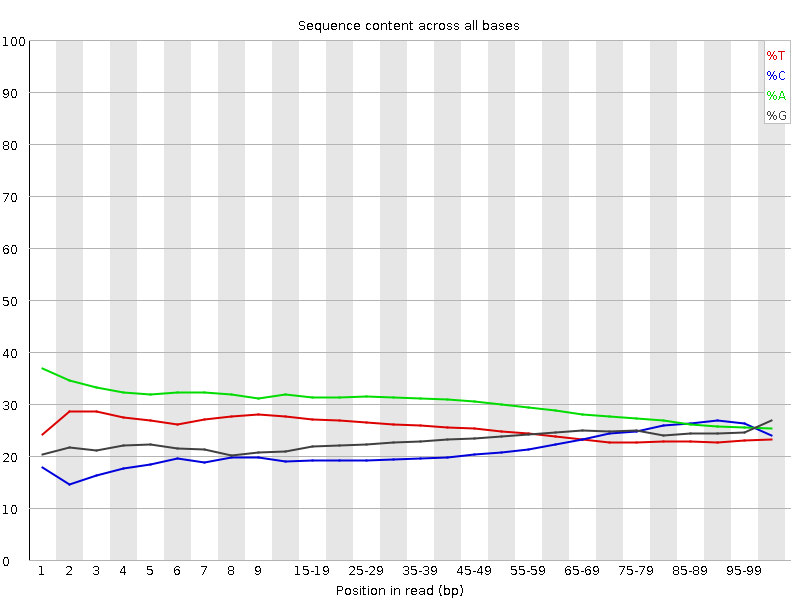
Is it possible you've just made a library with extremely short inserts and that this is why the quality is dropping so quickly? Given the adapter contamination you probably need to adapter trim your data anyway for it to be of any use, so you might as well quality trim while you're at it.Comment
-
The field of epigenetics has traditionally concentrated more on DNA and how changes like methylation and phosphorylation of histones impact gene expression and regulation. However, our increased understanding of RNA modifications and their importance in cellular processes has led to a rise in epitranscriptomics research. “Epitranscriptomics brings together the concepts of epigenetics and gene expression,” explained Adrien Leger, PhD, Principal Research Scientist...04-22-2024, 07:01 AM -
Proteins are often described as the workhorses of the cell, and identifying their sequences is key to understanding their role in biological processes and disease. Currently, the most common technique used to determine protein sequences is mass spectrometry. While still a valuable tool, mass spectrometry faces several limitations and requires a highly experienced scientist familiar with the equipment to operate it. Additionally, other proteomic methods, like affinity assays, are constrained...04-04-2024, 04:25 PM
Comment