 Tweet
Tweet
I wondering if there is any formula to estimate number of unique reads from FastQC Sequence Duplication Level output. For instance, in a set of 1M reads with 80% sequence duplication level what would be the estimated minimum and maximum of unique reads?
-
-
It would seem that the answer is 200,000 unique reads, but I don't know for sure.
You could download BBTools and run "dedupe.sh" on the dataset to get an exact number:
dedupe.sh in=reads.fq out=clean.fq
It works on single-ended or paired reads; for paired it only declares them duplicates if both reads match. Also, it supports variable #edits or substitutions, if you want, though by default it just looks for exact matches. -
Interpreting the FastQC duplication level plots is actually quite complicated.
See Simon Andrews' blog posts about this:
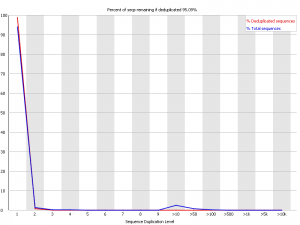
Comment
-
Thanks Brian and mastal for your comments and suggestions. I am only interested to know the number of unique reads from sequencing my libraries and I am reluctant to use any other tool since I get FastQC with other useful information. I wonder if someone could comment if a formula like this one: "(1-Sequence Duplication level%) x total number of reads= # of unique reads" will give correct answer or should some coefficients be factored in the formula.Comment
-
What I meant was, you can just run dedupe once on one dataset to confirm that the formula "(1-Sequence Duplication level%) x total number of reads= # of unique reads" is correct, or possibly derive a different formula, then go back to using FastQC. Dedupe prints the exact number (and percent) of duplicates.Comment

-
Proteins are often described as the workhorses of the cell, and identifying their sequences is key to understanding their role in biological processes and disease. Currently, the most common technique used to determine protein sequences is mass spectrometry. While still a valuable tool, mass spectrometry faces several limitations and requires a highly experienced scientist familiar with the equipment to operate it. Additionally, other proteomic methods, like affinity assays, are constrained...04-04-2024, 04:25 PM -
Despite advancements in sequencing platforms and related sample preparation technologies, certain sample types continue to present significant challenges that can compromise sequencing results. Pedro Echave, Senior Manager of the Global Business Segment at Revvity, explained that the success of a sequencing experiment ultimately depends on the amount and integrity of the nucleic acid template (RNA or DNA) obtained from a sample. “The better the quality of the nucleic acid isolated...03-22-2024, 06:39 AM
Comment