 Tweet
Tweet
Hi everyone,
I am using publicly available, 51 bp paired-end RNA seq data and I have some questions concerning the quality trimming of the data before passing them to Tophat2 for mapping.
Specifically I do not know which adapters were used, so I used fastqc and then trim_galore to remove the default illumina adapter "AGATCGGAAGAGC" and one overrepresented sequence "CTTTGTGTTTGATTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT".
It is really important to remove as much adapter contamination as possible because my analysis has to do with discovering variations that may correspond to RNA editing, rather than studying gene expression.
So my questions are:
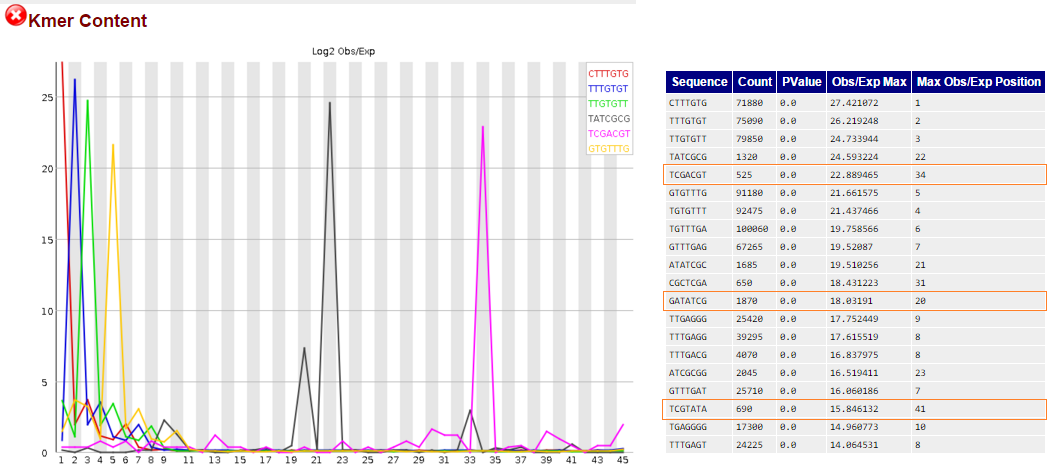
1) I am still getting 3 kmers in numbers ranging from 500 to 1800 that can be found within illumina adapters, and are reported to be in the middle of the read's length (positions 20, 34 and 41). Each one is found in a different adapter and an RNA PCR Index primer.
Should I use trim_galore to remove these kmers from my reads?
kmers in fastqc found in illumina adapters, marked in red boxes:
2) I have already performed removal with trim_galore for these kmers and trimming to improve Per base sequence content.
However the kmer GTACGTA appears in my fastqc report, and this kmer can be found in the TruSeq Adapter, Index 22. This adapter begins with GATCGGAAGAGC and should have been removed during the first step of trim_galore --illumina.
Should this kmer be removed as well?
Generally is it possible for kmers to be found within illumina adapters by chance?
3) After applying trim_galore --illumina the Per base sequence content of the 3 prime end of the reads is starting to show divergence, which gets worse every time I remove a sequence.
Is this because of the different length of the reads because some are trimmed more than others? (read length 51 to read length 20-51)
Should I trim the 3 prime end of the reads in this case?
Data before trimming: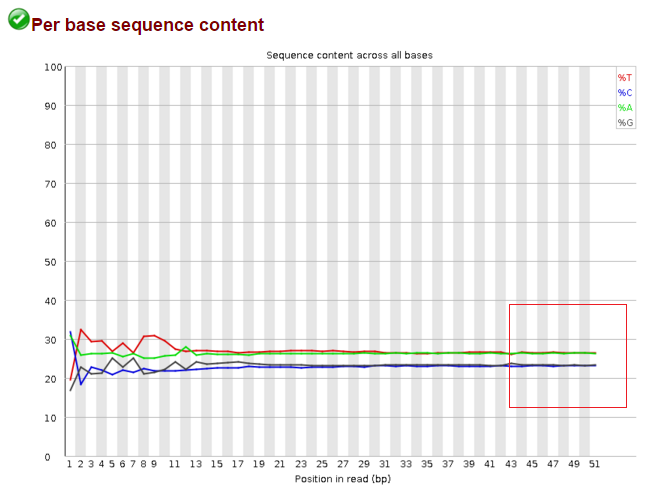
Data after trim_galore --illumina: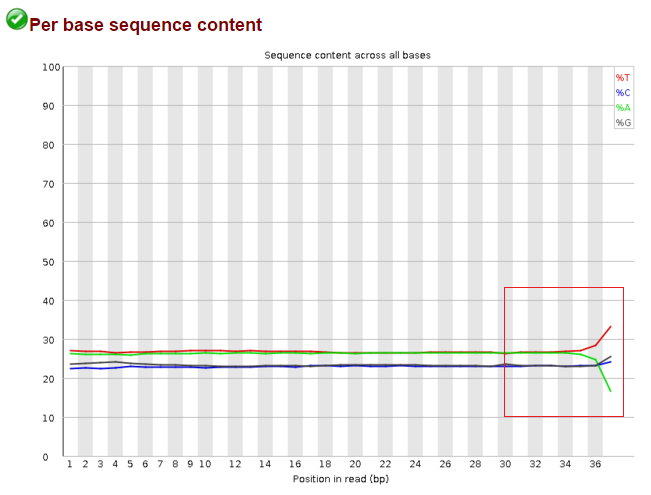
Data after trim_galore a)--illumina, b)kmers and c)overrexpressed sequence: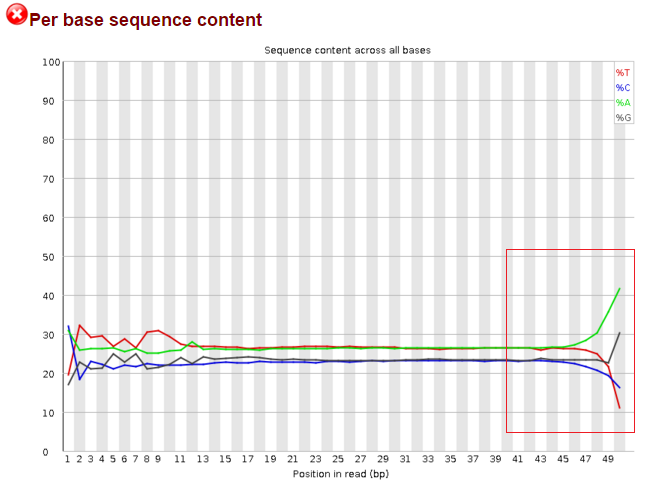
4) My last question is: Is tophat2 going to have a problem in alighning paired end reads with length ranging from 20 to 32?
I am using publicly available, 51 bp paired-end RNA seq data and I have some questions concerning the quality trimming of the data before passing them to Tophat2 for mapping.
Specifically I do not know which adapters were used, so I used fastqc and then trim_galore to remove the default illumina adapter "AGATCGGAAGAGC" and one overrepresented sequence "CTTTGTGTTTGATTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT".
It is really important to remove as much adapter contamination as possible because my analysis has to do with discovering variations that may correspond to RNA editing, rather than studying gene expression.
So my questions are:
1) I am still getting 3 kmers in numbers ranging from 500 to 1800 that can be found within illumina adapters, and are reported to be in the middle of the read's length (positions 20, 34 and 41). Each one is found in a different adapter and an RNA PCR Index primer.
Should I use trim_galore to remove these kmers from my reads?
kmers in fastqc found in illumina adapters, marked in red boxes:
2) I have already performed removal with trim_galore for these kmers and trimming to improve Per base sequence content.
However the kmer GTACGTA appears in my fastqc report, and this kmer can be found in the TruSeq Adapter, Index 22. This adapter begins with GATCGGAAGAGC and should have been removed during the first step of trim_galore --illumina.
Should this kmer be removed as well?
Generally is it possible for kmers to be found within illumina adapters by chance?
3) After applying trim_galore --illumina the Per base sequence content of the 3 prime end of the reads is starting to show divergence, which gets worse every time I remove a sequence.
Is this because of the different length of the reads because some are trimmed more than others? (read length 51 to read length 20-51)
Should I trim the 3 prime end of the reads in this case?
Data before trimming:
Data after trim_galore --illumina:
Data after trim_galore a)--illumina, b)kmers and c)overrexpressed sequence:
4) My last question is: Is tophat2 going to have a problem in alighning paired end reads with length ranging from 20 to 32?
Comment