 Tweet
Tweet
Hi all,
I have a collection of SNP data from a tetraploid population, and I would like to calculate the nucleotide diversity. The generel forumula is:
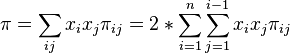
where xi and xj are the respective frequencies of the ith and jth sequences, (PI)ij is the number of nucleotide differences per nucleotide site between the ith and jth sequences, and n is the number of sequences in the sample.
This is fairly simple if we have sequence data (and not SNP data).
If we have these 3 sequences in a (very small) population, and we have sampled them each 1 time, we need to compare 1vs2, 1vs3, and 2vs3
>1
ATGCGTTTTT
>2
ATGGGTTTTT
>3
ATGCGTTTTA
We now get:
(PI)12 = 1/10 (they differ at pos. 4)
(PI)13 = 1/10 (they differ at pos. 10)
(PI)23 = 2/10 (they differ at pos. 4 and 10)
n = 3
The respective frequencies are always 1/3 in this case this means that:
PI = 2* ( (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(2/10) )
=>
PI = 2*(1/3^2)*(1/10+1/10+2/10)
=>
PI = (2/9)*(2/5)
=>
PI = 4/45 = 0.0888
I have a (VERY small popoluation of 2), and I have measured 2 SNPs:
Individual 1: a G/C SNP at position 3.
Individual 2: a T/A SNP at position 10.
Lets say the reference sequence is:
>ref
ATGCGTTTTT
Q1: Now I want to calculate the nucleotide diversity, would it then be correct to state that I have actually 4 sequences (4 alleles), meaning that I can calculate PI as follows:
Based on the SNP information:
>1 Individual 1 - allele 1
ATGCGTTTTT
>2 Individual 1 - allele 2
ATCCGTTTTT
>3 Individual 2 - allele 1
ATGCGTTTTT
>4 Individual 2 - allele 2
ATGCGTTTTA
We now get:
(PI)12 = 1/10 (they differ at pos. 3)
(PI)13 = 0 (they do not differ)
(PI)14 = 1/10 (they differ at pos. 10)
(PI)23 = 1/10 (they differ at pos. 3)
(PI)24 = 2/10 (they differ at pos. 3 and 10)
(PI)34 = 1/10 (they differ at pos. 10)
n = 4
The respective frequencies are always 1/4, and can be moved out of the parenthesis and squared (see above). in this case PI is:
PI = 2* ( (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(2/10) )
=>
PI = 2*(1/4^2)*(1/10+0+1/10+1/10+2/10+1/10)
=>
PI = (2/16)*(5/10)
=>
PI = 1/16
The question is whether this splitting of SNP information into alleles, and hence sequences is correct?
Now, as I stated I have a tetraploid population, and I therefore plan to do the same, but expand each SNP into 4 alleles:
I have a (VERY small population of 2), and I have measured 2 SNPs:
Individual 1: a G/G/C/C SNP at position 3.
Individual 2: a T/T/T/A SNP at position 10.
Lets say the reference sequence is still:
>ref
ATGCGTTTTT
Based on the SNP information:
>1 Individual 1 - allele 1
ATGCGTTTTT
>2 Individual 1 - allele 2
ATGCGTTTTT
>3 Individual 1 - allele 3
ATCCGTTTTT
>4 Individual 1 - allele 4
ATCCGTTTTT
>5 Individual 2 - allele 1
ATGCGTTTTT
>6 Individual 2 - allele 2
ATGCGTTTTT
>7 Individual 2 - allele 3
ATGCGTTTTT
>8 Individual 2 - allele 4
ATGCGTTTTA
n = 8
(PI)12 = 0 (they do not differ)
(PI)13 = 1/10 (they differ at pos. 3)
(PI)14 = 1/10 (they differ at pos. 3)
(PI)15 = 0 (they do not differ)
(PI)16 = 0 (they do not differ)
(PI)17 = 0 (they do not differ)
(PI)18 = 1/10 (they differ at pos. 10)
(PI)23 = 1/10 (they differ at pos. 3)
(PI)24 = 1/10 (they differ at pos. 3)
(PI)25 = 0 (they do not differ)
(PI)26 = 0 (they do not differ)
(PI)27 = 0 (they do not differ)
(PI)28 = 1/10 (they differ at pos. 10)
(PI)34 = 0 (they do not differ)
(PI)35 = 1/10 (they differ at pos. 3)
(PI)36 = 1/10 (they differ at pos. 3)
(PI)37 = 1/10 (they differ at pos. 3)
(PI)38 = 2/10 (they differ at pos. 3 and 10)
(PI)45 = 1/10 (they differ at pos. 3)
(PI)46 = 1/10 (they differ at pos. 3)
(PI)47 = 1/10 (they differ at pos. 3)
(PI)48 = 2/10 (they differ at pos. 3 and 10)
(PI)56 = 0 (they do not differ)
(PI)57 = 0 (they do not differ)
(PI)58 = 1/10 (they differ at pos. 10)
(PI)67 = 0 (they do not differ)
(PI)68 = 1/10 (they differ at pos. 10)
(PI)78 = 1/10 (they differ at pos. 10)
Now, the respective frequencies are always 1/8, and can be moved out of the parenthesis and squared (see above). in this case PI is:
SUM( PI(XJ) ) = 19/10
PI = 2* ( (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(2/10) )
=>
PI = 2*(1/8^2)*(19/10)
=>
PI = (1/32)*(19/10)
=>
PI = 19/320 = 0.059375
The question is (again) whether this splitting of SNP information into alleles, and hence sequences is correct?
I have a collection of SNP data from a tetraploid population, and I would like to calculate the nucleotide diversity. The generel forumula is:
where xi and xj are the respective frequencies of the ith and jth sequences, (PI)ij is the number of nucleotide differences per nucleotide site between the ith and jth sequences, and n is the number of sequences in the sample.
This is fairly simple if we have sequence data (and not SNP data).
If we have these 3 sequences in a (very small) population, and we have sampled them each 1 time, we need to compare 1vs2, 1vs3, and 2vs3
>1
ATGCGTTTTT
>2
ATGGGTTTTT
>3
ATGCGTTTTA
We now get:
(PI)12 = 1/10 (they differ at pos. 4)
(PI)13 = 1/10 (they differ at pos. 10)
(PI)23 = 2/10 (they differ at pos. 4 and 10)
n = 3
The respective frequencies are always 1/3 in this case this means that:
PI = 2* ( (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(2/10) )
=>
PI = 2*(1/3^2)*(1/10+1/10+2/10)
=>
PI = (2/9)*(2/5)
=>
PI = 4/45 = 0.0888
I have a (VERY small popoluation of 2), and I have measured 2 SNPs:
Individual 1: a G/C SNP at position 3.
Individual 2: a T/A SNP at position 10.
Lets say the reference sequence is:
>ref
ATGCGTTTTT
Q1: Now I want to calculate the nucleotide diversity, would it then be correct to state that I have actually 4 sequences (4 alleles), meaning that I can calculate PI as follows:
Based on the SNP information:
>1 Individual 1 - allele 1
ATGCGTTTTT
>2 Individual 1 - allele 2
ATCCGTTTTT
>3 Individual 2 - allele 1
ATGCGTTTTT
>4 Individual 2 - allele 2
ATGCGTTTTA
We now get:
(PI)12 = 1/10 (they differ at pos. 3)
(PI)13 = 0 (they do not differ)
(PI)14 = 1/10 (they differ at pos. 10)
(PI)23 = 1/10 (they differ at pos. 3)
(PI)24 = 2/10 (they differ at pos. 3 and 10)
(PI)34 = 1/10 (they differ at pos. 10)
n = 4
The respective frequencies are always 1/4, and can be moved out of the parenthesis and squared (see above). in this case PI is:
PI = 2* ( (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(2/10) )
=>
PI = 2*(1/4^2)*(1/10+0+1/10+1/10+2/10+1/10)
=>
PI = (2/16)*(5/10)
=>
PI = 1/16
The question is whether this splitting of SNP information into alleles, and hence sequences is correct?
Now, as I stated I have a tetraploid population, and I therefore plan to do the same, but expand each SNP into 4 alleles:
I have a (VERY small population of 2), and I have measured 2 SNPs:
Individual 1: a G/G/C/C SNP at position 3.
Individual 2: a T/T/T/A SNP at position 10.
Lets say the reference sequence is still:
>ref
ATGCGTTTTT
Based on the SNP information:
>1 Individual 1 - allele 1
ATGCGTTTTT
>2 Individual 1 - allele 2
ATGCGTTTTT
>3 Individual 1 - allele 3
ATCCGTTTTT
>4 Individual 1 - allele 4
ATCCGTTTTT
>5 Individual 2 - allele 1
ATGCGTTTTT
>6 Individual 2 - allele 2
ATGCGTTTTT
>7 Individual 2 - allele 3
ATGCGTTTTT
>8 Individual 2 - allele 4
ATGCGTTTTA
n = 8
(PI)12 = 0 (they do not differ)
(PI)13 = 1/10 (they differ at pos. 3)
(PI)14 = 1/10 (they differ at pos. 3)
(PI)15 = 0 (they do not differ)
(PI)16 = 0 (they do not differ)
(PI)17 = 0 (they do not differ)
(PI)18 = 1/10 (they differ at pos. 10)
(PI)23 = 1/10 (they differ at pos. 3)
(PI)24 = 1/10 (they differ at pos. 3)
(PI)25 = 0 (they do not differ)
(PI)26 = 0 (they do not differ)
(PI)27 = 0 (they do not differ)
(PI)28 = 1/10 (they differ at pos. 10)
(PI)34 = 0 (they do not differ)
(PI)35 = 1/10 (they differ at pos. 3)
(PI)36 = 1/10 (they differ at pos. 3)
(PI)37 = 1/10 (they differ at pos. 3)
(PI)38 = 2/10 (they differ at pos. 3 and 10)
(PI)45 = 1/10 (they differ at pos. 3)
(PI)46 = 1/10 (they differ at pos. 3)
(PI)47 = 1/10 (they differ at pos. 3)
(PI)48 = 2/10 (they differ at pos. 3 and 10)
(PI)56 = 0 (they do not differ)
(PI)57 = 0 (they do not differ)
(PI)58 = 1/10 (they differ at pos. 10)
(PI)67 = 0 (they do not differ)
(PI)68 = 1/10 (they differ at pos. 10)
(PI)78 = 1/10 (they differ at pos. 10)
Now, the respective frequencies are always 1/8, and can be moved out of the parenthesis and squared (see above). in this case PI is:
SUM( PI(XJ) ) = 19/10
PI = 2* ( (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(1/10) + (1/3)*(1/3)*(2/10) )
=>
PI = 2*(1/8^2)*(19/10)
=>
PI = (1/32)*(19/10)
=>
PI = 19/320 = 0.059375
The question is (again) whether this splitting of SNP information into alleles, and hence sequences is correct?
 ? Another case is, when there are C to A mutation, in another place we found A deletion. Its like an equilibrium to balancq A composition. Any one who can explain this phenomenon? Thank you All
? Another case is, when there are C to A mutation, in another place we found A deletion. Its like an equilibrium to balancq A composition. Any one who can explain this phenomenon? Thank you All
Comment