 Tweet
Tweet
Hi,
In my center we are interested in calling CNVs from targeted resequencing using Illumina's MiSeq and I am facing the following issue:
Two of the regions of interest share high homology, thus, creating multimapped reads when aligning reads of 76 bps. We are currently using two mappers, BWA MEM (default parameters), and GEM (1% mismatch).
When extracting reads from this regions I retrieve nearly the half coverage using BWA MEM, compared to GEM. Correct me if I am wrong, but I understand that BWA MEM chooses a secondary hit at random, whereas GEM reports every location where the read aligns. This shouldn't be an issue for CNV calling after normalizing the coverage and computing ratios between a pool of samples, but the problem comes when a sample has two consecutive SNPs in the region (image attached).
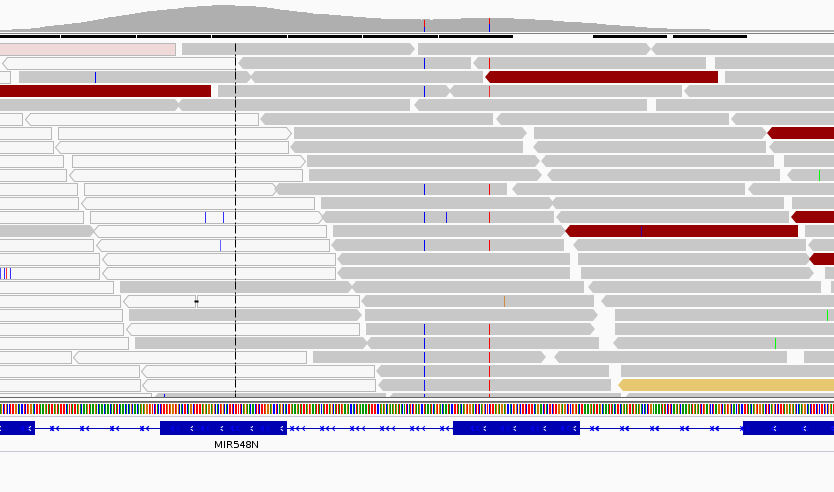
In this scenario, I get more coverage for the samples with the SNPs compared to the others when aligning with BWA MEM, but I don't see any effect when mapping with GEM. Finally this produces a false positive when calling a CNV. So I am trying to understand why this could be happening. My guess is that this SNPs are increasing the mappability of this reads (helped by mate rescue?), so BWA MEM produces less multimapped reads.
Does anybody know what bwa mem does here?
In my center we are interested in calling CNVs from targeted resequencing using Illumina's MiSeq and I am facing the following issue:
Two of the regions of interest share high homology, thus, creating multimapped reads when aligning reads of 76 bps. We are currently using two mappers, BWA MEM (default parameters), and GEM (1% mismatch).
When extracting reads from this regions I retrieve nearly the half coverage using BWA MEM, compared to GEM. Correct me if I am wrong, but I understand that BWA MEM chooses a secondary hit at random, whereas GEM reports every location where the read aligns. This shouldn't be an issue for CNV calling after normalizing the coverage and computing ratios between a pool of samples, but the problem comes when a sample has two consecutive SNPs in the region (image attached).
In this scenario, I get more coverage for the samples with the SNPs compared to the others when aligning with BWA MEM, but I don't see any effect when mapping with GEM. Finally this produces a false positive when calling a CNV. So I am trying to understand why this could be happening. My guess is that this SNPs are increasing the mappability of this reads (helped by mate rescue?), so BWA MEM produces less multimapped reads.
Does anybody know what bwa mem does here?