 Tweet
Tweet
Hi @all,
we are currently struggling with pros/cons of buying a PB sequel vs ONT Promethion for our analyses (human de novo single celll WGS). Ideally we are looking for something with which we can analyse structural variants and SNPs (so something with ~100Gb+ (>30x) output).
Unfortunately, we're getting extremely diverse information about the specifications of the two systems. Hence, I'd like to ask users of each system for info on their experience (I have therefore a similar post in the pacbio forum).
My information on the promethion so far:
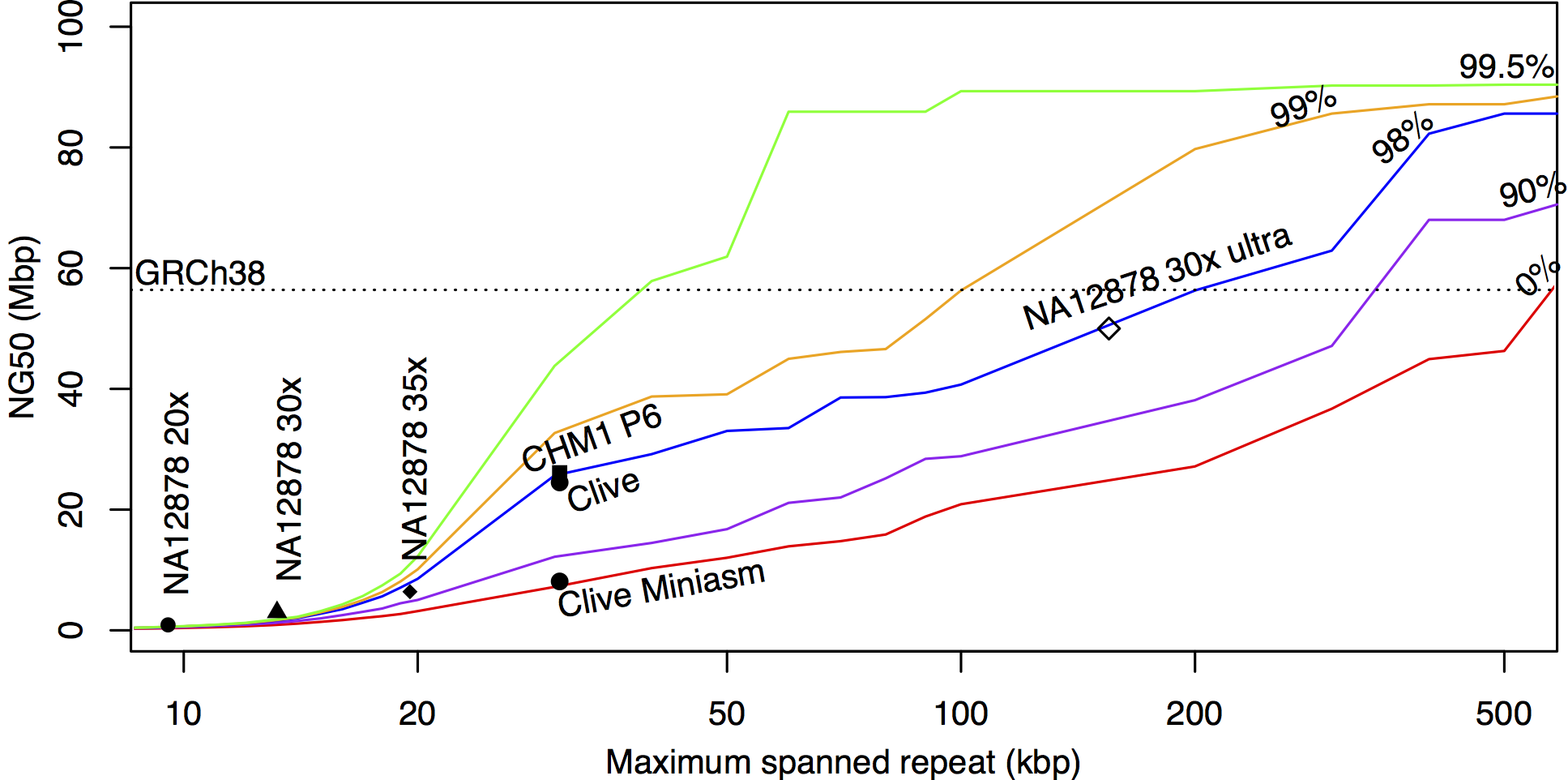
1) Accuracy of >95%
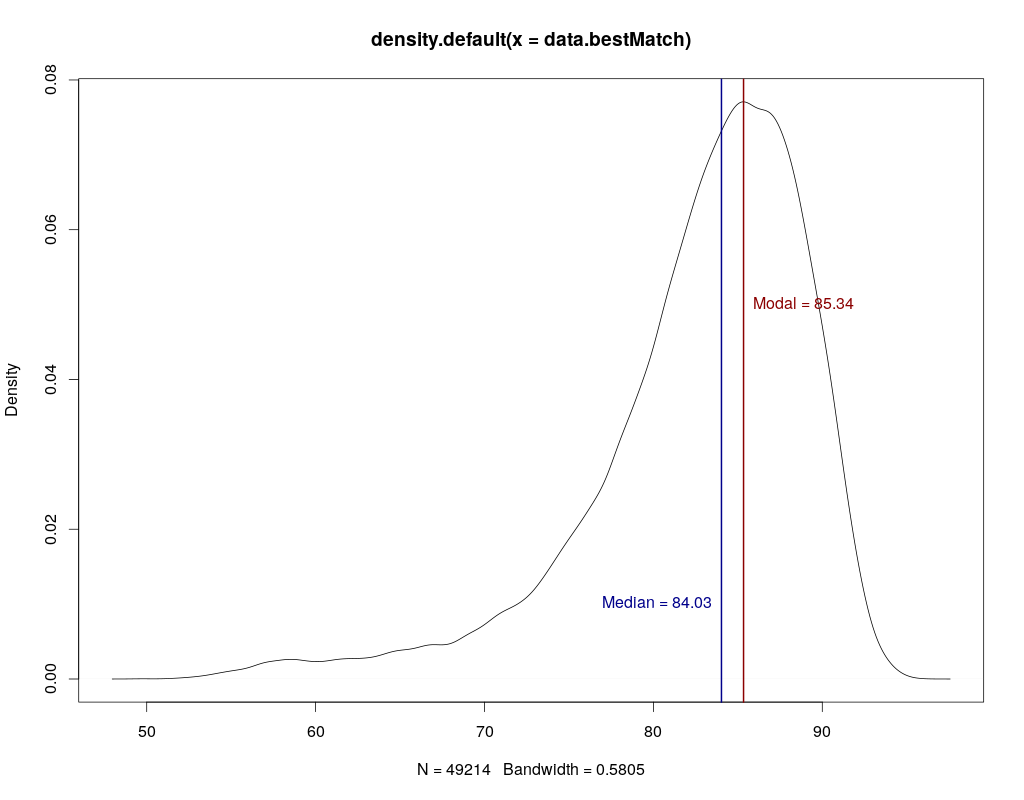
Is this a consensus acc from assembly/mapping? As this would be coverage dependent, it is meaningless for me... What about individual reads (1D or 2D) accuracy? I have looked briefly into the available data on github which looks more like 50-70% acc for single reads (but I might just have been unlucky with my selected genomic regions...)
2) I got information of 3-11Gb per flow cell / ~900€ per flow cell
So, I would need 9-33 flow cells / genome => ~8k - 30k€ / genome?
3) Price ~150k €
Is installation, training, etc. included?
4) New developments
I'm a little concerned with the fast release of new protocols and chemistries. Although I understand that this is a system under development, how big are the differences between new releases? It seems to my like every tiny new improvement a new release?
I would be grateful for any information on any of my four points.
As minion and promethion use the same pore and same chemistry, information on both would be appreciated.
Thanks!
we are currently struggling with pros/cons of buying a PB sequel vs ONT Promethion for our analyses (human de novo single celll WGS). Ideally we are looking for something with which we can analyse structural variants and SNPs (so something with ~100Gb+ (>30x) output).
Unfortunately, we're getting extremely diverse information about the specifications of the two systems. Hence, I'd like to ask users of each system for info on their experience (I have therefore a similar post in the pacbio forum).
My information on the promethion so far:
1) Accuracy of >95%
Is this a consensus acc from assembly/mapping? As this would be coverage dependent, it is meaningless for me... What about individual reads (1D or 2D) accuracy? I have looked briefly into the available data on github which looks more like 50-70% acc for single reads (but I might just have been unlucky with my selected genomic regions...)
2) I got information of 3-11Gb per flow cell / ~900€ per flow cell
So, I would need 9-33 flow cells / genome => ~8k - 30k€ / genome?
3) Price ~150k €
Is installation, training, etc. included?
4) New developments
I'm a little concerned with the fast release of new protocols and chemistries. Although I understand that this is a system under development, how big are the differences between new releases? It seems to my like every tiny new improvement a new release?
I would be grateful for any information on any of my four points.
As minion and promethion use the same pore and same chemistry, information on both would be appreciated.
Thanks!



Comment