 Tweet
Tweet
Method:
Wang et al. 2011, A low-cost library construction protocol for Illumina-based strand-specific multiplex rna-seq. PLoS One 6(10) e26426. doi:10.1371/journal.pone.0026426
Basics steps:
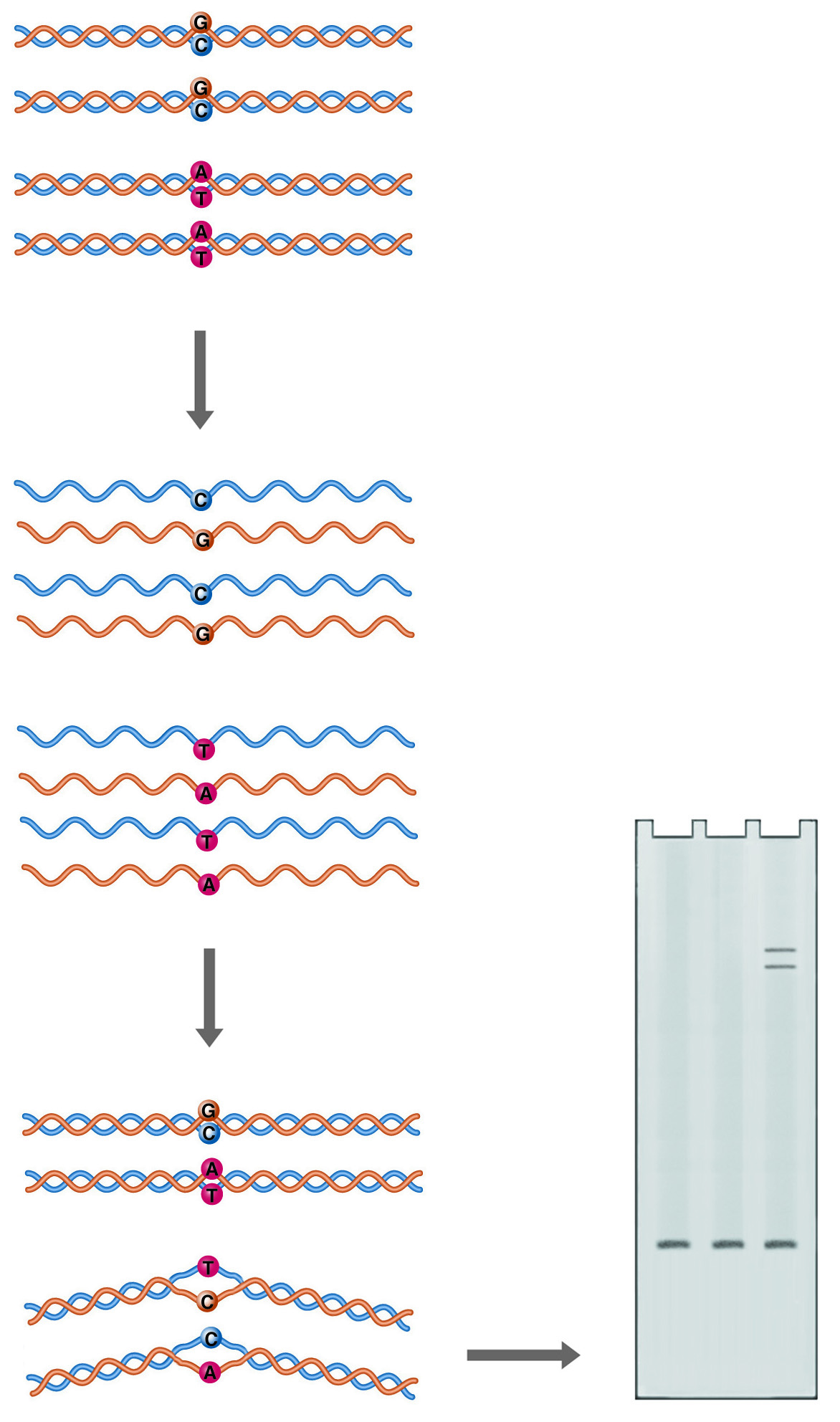
Extract Total RNA
Extract mRNA
Fragment
1st strand synthesis
2nd strand synthesis
End repair
dA-Tail
Ligate adaptors
Size select
Excise dUTP
Amplify
Bioanalyze with DNA HS Chip
Problem:
Bioanalysis with a DNA HS Chip, reveals a double hump in every library product.

Notes:
Libraries are individual i.e. not yet pooled.
Yes Library 11 has a ~126bp adaptor dimer (already fixed).
100ng mRNA input
Qubit after cDNA synthesis (2nd strand) yields on average 8-10ng total
Qubit after final library completion, anything from 8-30ng total from 14 cycles.
Amplification step is definitely working (trace not shown).
I made many more libraries with this method and the double humps just keep on coming.
Questions:
1. What is happening here, any theories?
2. How do I fix this?
EDIT - 26 Nov 13
Guys I don't believe the double hump problems are related to the following examples:

PCR artifacts are normally twice the expected library size as shown in the image above. Since my libraries are between 200-500bp which is exactly the expected size I don't believe my results are PCR artifact related, but I'm happy to be proven wrong. Notice in my samples the double humps are within the main library peak and not two separate peaks.

There is definitely no indication of bead carry over in my traces.
Things I have tried but made no difference:
• Denaturing the libraries multiple different ways to prevent bird nesting or this so called “bubble effect” as suggested in other posts
• I have tried re-amplifying my libraries for another 2 more cycles as suggest in other posts
• Tried 12x, 13x, 14x, 15x, 16x, 17x cycles.
Wang et al. 2011, A low-cost library construction protocol for Illumina-based strand-specific multiplex rna-seq. PLoS One 6(10) e26426. doi:10.1371/journal.pone.0026426
Basics steps:
Extract Total RNA
Extract mRNA
Fragment
1st strand synthesis
2nd strand synthesis
End repair
dA-Tail
Ligate adaptors
Size select
Excise dUTP
Amplify
Bioanalyze with DNA HS Chip
Problem:
Bioanalysis with a DNA HS Chip, reveals a double hump in every library product.
Notes:
Libraries are individual i.e. not yet pooled.
Yes Library 11 has a ~126bp adaptor dimer (already fixed).
100ng mRNA input
Qubit after cDNA synthesis (2nd strand) yields on average 8-10ng total
Qubit after final library completion, anything from 8-30ng total from 14 cycles.
Amplification step is definitely working (trace not shown).
I made many more libraries with this method and the double humps just keep on coming.
Questions:
1. What is happening here, any theories?
2. How do I fix this?
EDIT - 26 Nov 13
Guys I don't believe the double hump problems are related to the following examples:
PCR artifacts are normally twice the expected library size as shown in the image above. Since my libraries are between 200-500bp which is exactly the expected size I don't believe my results are PCR artifact related, but I'm happy to be proven wrong. Notice in my samples the double humps are within the main library peak and not two separate peaks.
There is definitely no indication of bead carry over in my traces.
Things I have tried but made no difference:
• Denaturing the libraries multiple different ways to prevent bird nesting or this so called “bubble effect” as suggested in other posts
• I have tried re-amplifying my libraries for another 2 more cycles as suggest in other posts
• Tried 12x, 13x, 14x, 15x, 16x, 17x cycles.
Comment