 Tweet
Tweet
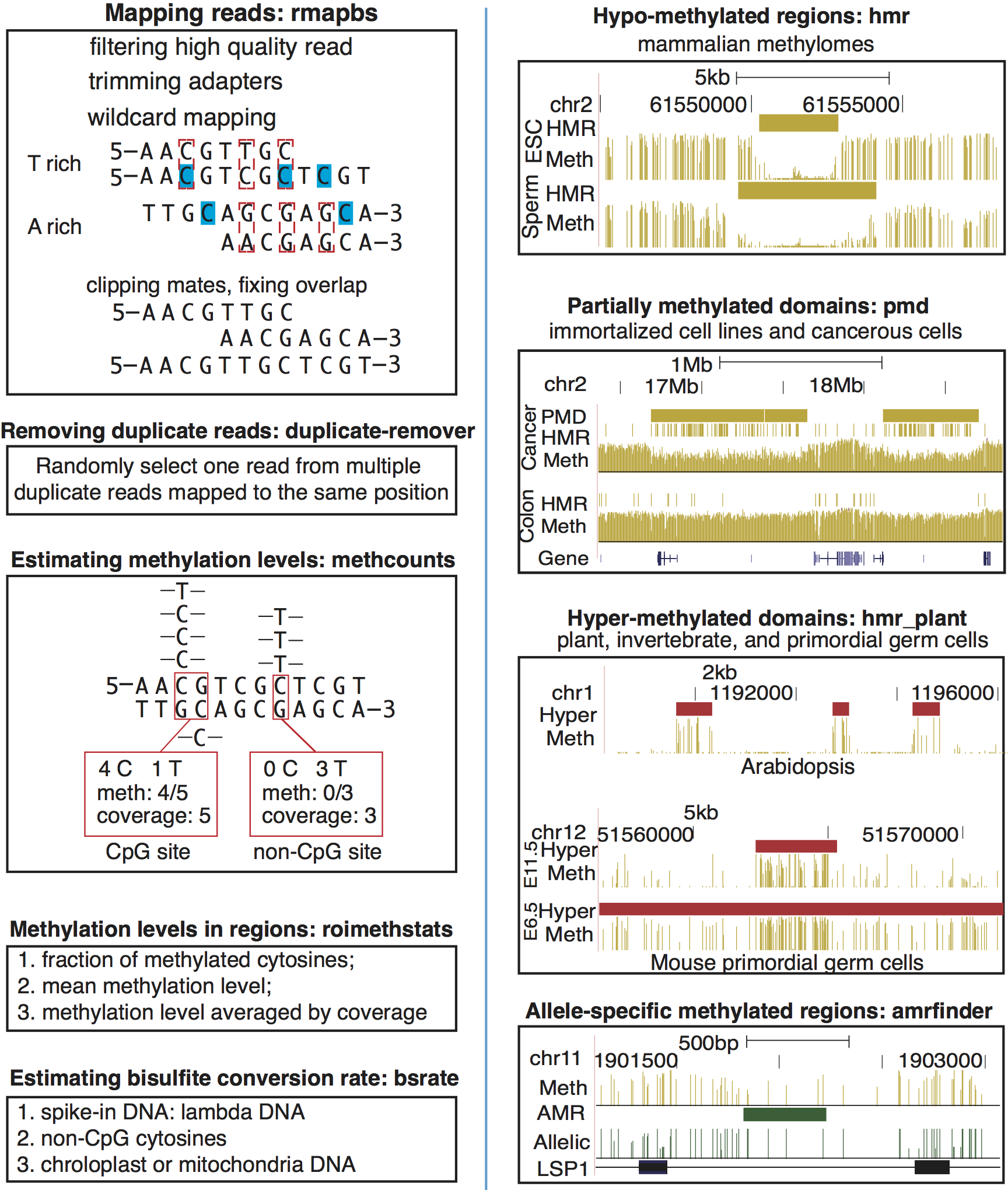
Software tools for comparing entire methylomes (WGBS)
I am currently exploring the possibility of comparing entire methylomes of genets exposed to two different conditions.
There seem to be a few options as for software tools to map bisulfite-seq reads (Bismark), but the options for comparing entire methylomes don't seem to exist as a tools as of yet.
Essentially, I would like to model the work of Heyn et al. 2012. The details of their methods are available in supplemental materials.
Is there a less cumbersome way of achieving what they are doing? Are there any tools capable of this? What is the most efficient way of identifying differential methylation between two samples?
Thank you!
I am currently exploring the possibility of comparing entire methylomes of genets exposed to two different conditions.
There seem to be a few options as for software tools to map bisulfite-seq reads (Bismark), but the options for comparing entire methylomes don't seem to exist as a tools as of yet.
Essentially, I would like to model the work of Heyn et al. 2012. The details of their methods are available in supplemental materials.
Is there a less cumbersome way of achieving what they are doing? Are there any tools capable of this? What is the most efficient way of identifying differential methylation between two samples?
Thank you!

Comment