 Tweet
Tweet
Hi,
I am using fastQC to check the quality of my miRNA seq data after adaptor trimming. Then I used fastq_quality_boxplot_graph.sh from fastx toolkits to see if the results are the same as fastQC. Surprisingly, it seems that with fastq_quality_boxplot_graph.sh, the quality scores are lower (some below 30) whereas they are all above 30 using fastQC.
Could anybody tell me why there is so big difference?
There is another thread:

But in my case, I believe that all the reads have been read by fastQC or fastq_quality_boxplot_graph.sh, which can be known from their statistics.
There is indeed a very small percentage of reads which contain N. Could that be the problem? I am looking for some tools to get rid of the reads with N.
Attached are the results from fastQC and fastq_quality_boxplot_graph.sh. Thanks.
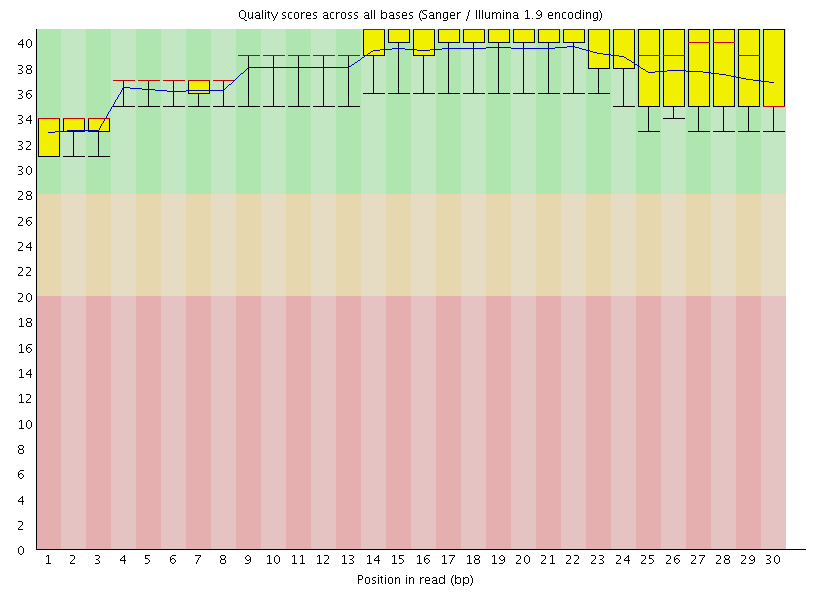
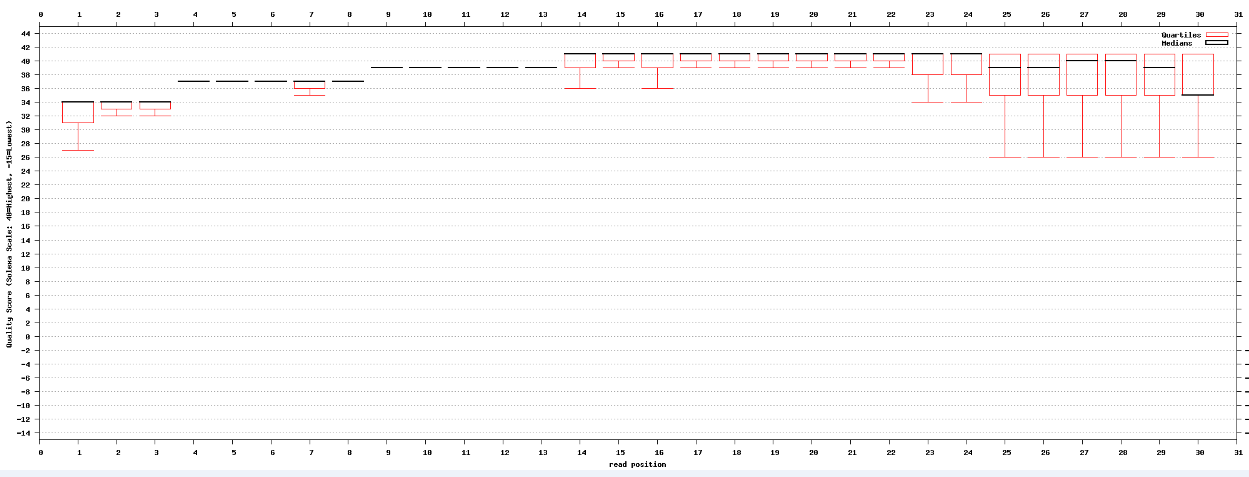
I am using fastQC to check the quality of my miRNA seq data after adaptor trimming. Then I used fastq_quality_boxplot_graph.sh from fastx toolkits to see if the results are the same as fastQC. Surprisingly, it seems that with fastq_quality_boxplot_graph.sh, the quality scores are lower (some below 30) whereas they are all above 30 using fastQC.
Could anybody tell me why there is so big difference?
There is another thread:
But in my case, I believe that all the reads have been read by fastQC or fastq_quality_boxplot_graph.sh, which can be known from their statistics.
There is indeed a very small percentage of reads which contain N. Could that be the problem? I am looking for some tools to get rid of the reads with N.
Attached are the results from fastQC and fastq_quality_boxplot_graph.sh. Thanks.
Comment