 Tweet
Tweet
Hi all, i am new to RNA-seq and i am using Galaxy to perform my analysis as part of a differential gene expression experiment. When using FASTQC to report on the quality of my data i see an over representation of the same k-mers ion the R2 reads of all my datasets. As you can see in the fastqc pictures the kmers GAAGA, GGAAG and AAGAG seem to be overepresented at the ends of my reads in the different datasets. I believe this may be indicative of adapter contamination but was unsure how to verify this and take steps to remove the contamination. Also can this contamination damage my TopHat alignments down the line.
Thanks
Richard

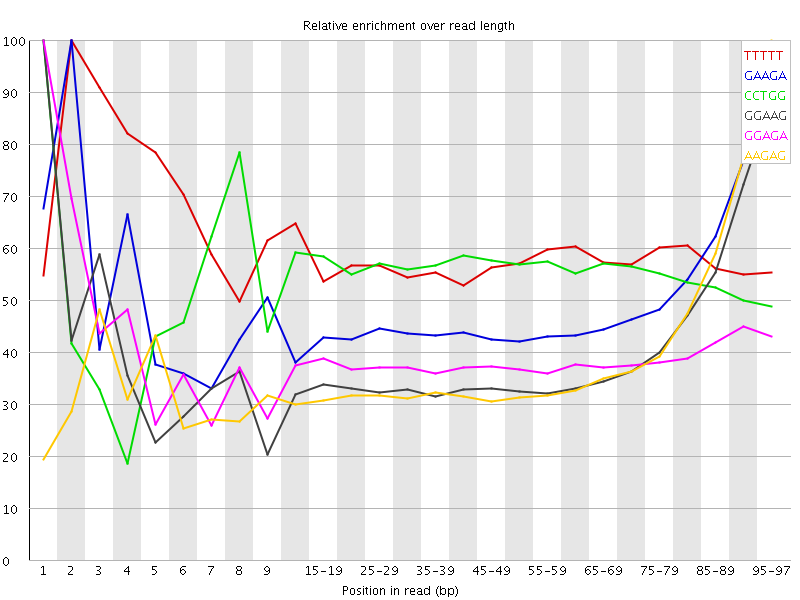
Thanks
Richard