 Tweet
Tweet
Hi,
I’m analyzing a metagenomics data set and am a bit concerned about the sequencing quality. It was run using NextSeq for PE150. I need a second opinion. Attached is Mothur's summary.seqs output from one of the samples, Sample10 as an example.
For the raw reads, the max read length is 151, the median is 129.
After assembly (stitch R1/R2 together, max is 292, median is 121.
Almost all the samples are like that more or less. Is this normal or is it an indication of poor seq quality?
thanks!
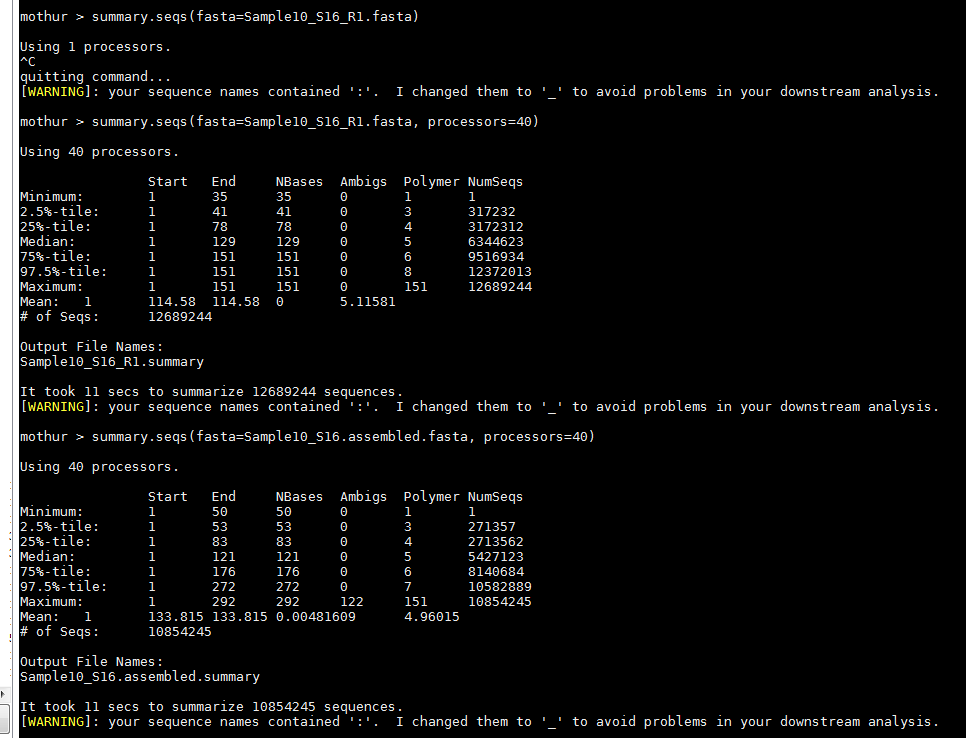
I’m analyzing a metagenomics data set and am a bit concerned about the sequencing quality. It was run using NextSeq for PE150. I need a second opinion. Attached is Mothur's summary.seqs output from one of the samples, Sample10 as an example.
For the raw reads, the max read length is 151, the median is 129.
After assembly (stitch R1/R2 together, max is 292, median is 121.
Almost all the samples are like that more or less. Is this normal or is it an indication of poor seq quality?
thanks!
Comment