 Tweet
Tweet
Hi everyone!
I'm trying to evaluate gene expression differences, in breast cancer cells before and after treatment. So I'm working on RNA-seq data (single-end reads).
I tried to correlate FPKM (CuffDiff output) and RPKM (counts from HTSeq-count, then classic "Mortazavi et al." calculation).
Reading the CuffLinks website, some papers and other forums, it seems that these values have to be the same for single-end reads data!
I also filtered miRNAs and other genes shorter then 300bp (could give false FPKM high values).
I hope that someone can help me!
Thanks in advace
I'm trying to evaluate gene expression differences, in breast cancer cells before and after treatment. So I'm working on RNA-seq data (single-end reads).
I tried to correlate FPKM (CuffDiff output) and RPKM (counts from HTSeq-count, then classic "Mortazavi et al." calculation).
Reading the CuffLinks website, some papers and other forums, it seems that these values have to be the same for single-end reads data!
I also filtered miRNAs and other genes shorter then 300bp (could give false FPKM high values).
I hope that someone can help me!
Thanks in advace
 My question is:
My question is: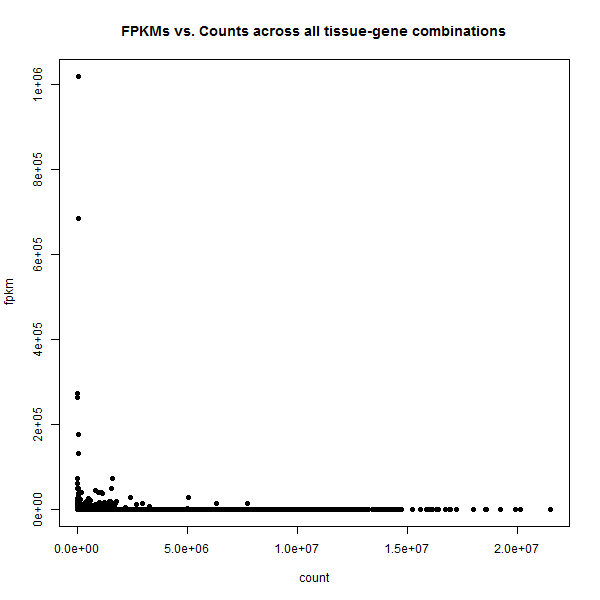

 I meant FPKM
I meant FPKM
Comment