 Tweet
Tweet
Previously I've used Illumina for my sequencing needs and recently I've been handed some Ion Torrent data to do RNA-Seq.
The fastqc results are significantly poorer than I'm used to. However, I do realise there is considerably different chemistry involved, and fastqc was designed for Illumina, so may not be giving accurate results.
Nevertheless, the large number of failed components is concerning and I was wondering if anyone experienced with Ion Torrent data can tell me if these results mean the samples need to be re-sequenced or not.

One of my main concerns, other than quality, is the variable size of the total number of sequences per sample. These vary from 12703948 sequences to 50092930 sequences. Is this normal for Ion Torrent? How can I accurately calculate differential expression with such variable sequences numbers per sample?
Filename 5C_IonXpressRNA_009_rawlib.basecaller.bam
File type Conventional base calls
Encoding Sanger / Illumina 1.9
Total Sequences 17356006
Sequences flagged as poor quality 0
Sequence length 8-352
%GC 48
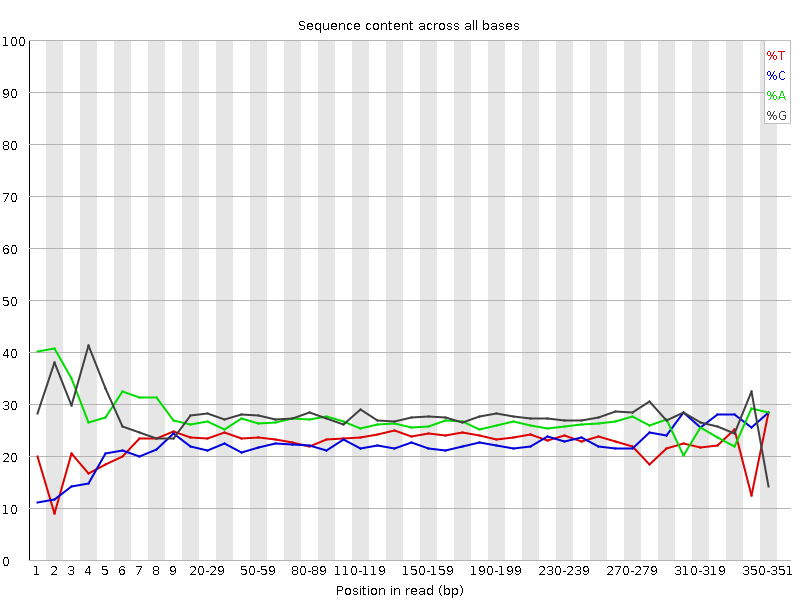
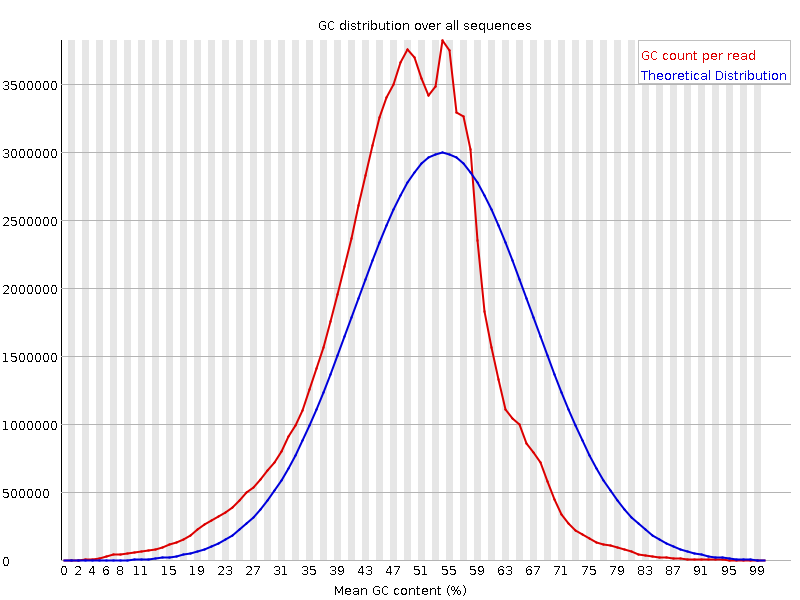
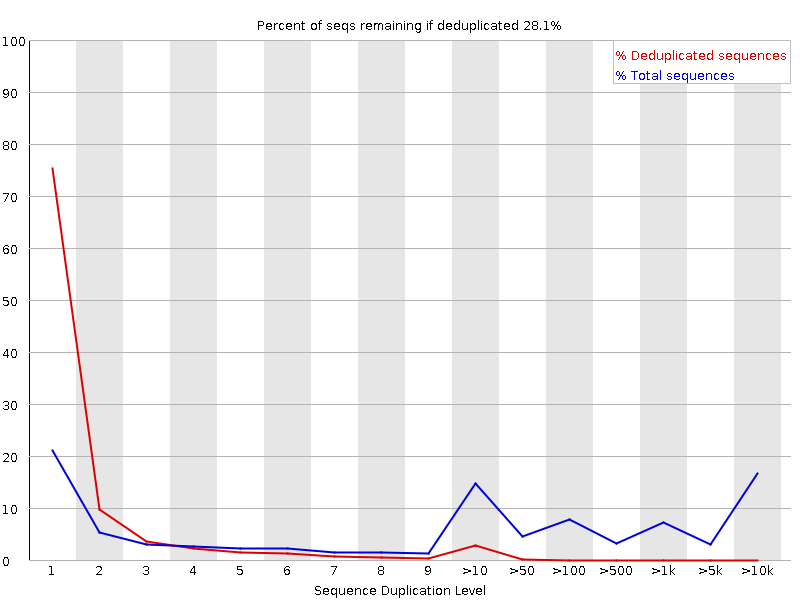
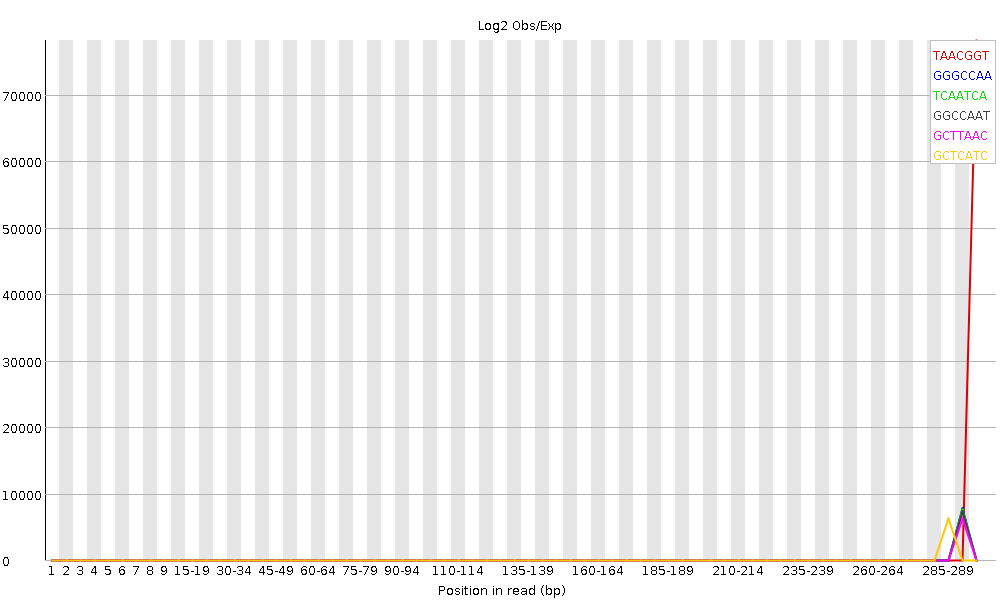
The fastqc results are significantly poorer than I'm used to. However, I do realise there is considerably different chemistry involved, and fastqc was designed for Illumina, so may not be giving accurate results.
Nevertheless, the large number of failed components is concerning and I was wondering if anyone experienced with Ion Torrent data can tell me if these results mean the samples need to be re-sequenced or not.
One of my main concerns, other than quality, is the variable size of the total number of sequences per sample. These vary from 12703948 sequences to 50092930 sequences. Is this normal for Ion Torrent? How can I accurately calculate differential expression with such variable sequences numbers per sample?
Filename 5C_IonXpressRNA_009_rawlib.basecaller.bam
File type Conventional base calls
Encoding Sanger / Illumina 1.9
Total Sequences 17356006
Sequences flagged as poor quality 0
Sequence length 8-352
%GC 48
Comment