 Tweet
Tweet
I'm trying to map some sRNA reads to specific loci of interest while allowing up to 3 mismatches using -v 3.
For some of my target loci, this seems to work well. All of the mapped reads have no more than 3 mismatches.
However, when I use the exact same code, changing only the bowtie files for a new locus, I'll get reads with up to 20 mismatches (and the sRNA read is only 24nt long!).
I've attached two screenshots from IGV showing the same sRNA sample mapped with Bowtie against two different loci. All of the nucleotides shown are mismatches.
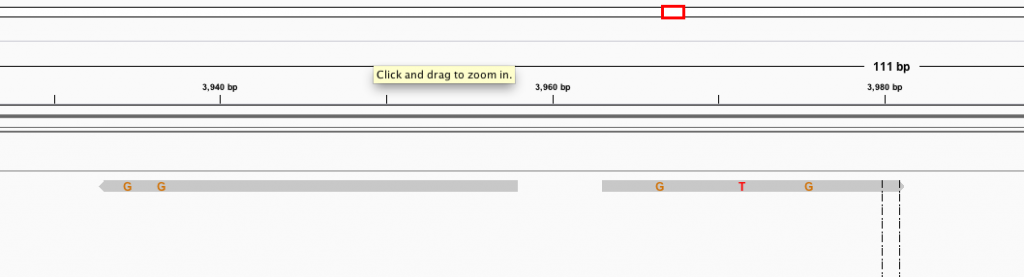
How I expect it to always look:
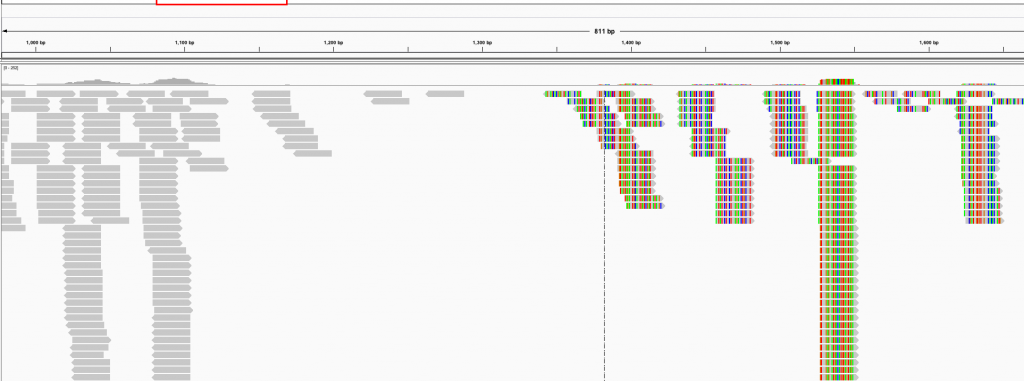
How it usually turns out:

The code I'm using is as follows:
Oddly, I also have the exact same issue (too many mismatches) when I use this code:
Am I doing something wrong? Has anyone else seen this?
For some of my target loci, this seems to work well. All of the mapped reads have no more than 3 mismatches.
However, when I use the exact same code, changing only the bowtie files for a new locus, I'll get reads with up to 20 mismatches (and the sRNA read is only 24nt long!).
I've attached two screenshots from IGV showing the same sRNA sample mapped with Bowtie against two different loci. All of the nucleotides shown are mismatches.
How I expect it to always look:
How it usually turns out:
The code I'm using is as follows:
Code:
bowtie -f -v 3 -S ~/CRM2 ~/sRNA.fa > ~/output.sam
Code:
bowtie -f -n 0 -l 25 -S ~/CRM2 ~/sRNA.fa > ~/output.sam

Comment