 Tweet
Tweet
Hello experts,
I am following the recently published Nature Protocol guideline for analyzing my RNA-seq runs using tophat-cufflink-cuffmerge-cuffdiff (cufflink.1.3.0). I wanted see whether upper quantile normalization improved my results so I also used the "-N" option for Cuffdiff. But Here are some summaries from the "gene_exp.diff" output files with or without the "-N" option:
Condition #ok.status Correlation #Significant DE.Sample.1>Sample.2 DE.Sample.2>sample.1
Without -N 11884 0.6156752 3865 1851 2014
With -N 11885 0.6139065 4435 1620 2815
My questions are:
1. Is it common to see such HUGE variations on the number of significantly DE genes, the numbers of DE sample 1 > sample 2 and sample 2 > sample 1 just for adding this "-N" option?? and since there's such a huge change, which DE gene list should I trust??
2. I noticed there is also "-N" option for cufflink. So at which step should I use such option, and will using "--total-hits-norm" also advisable?
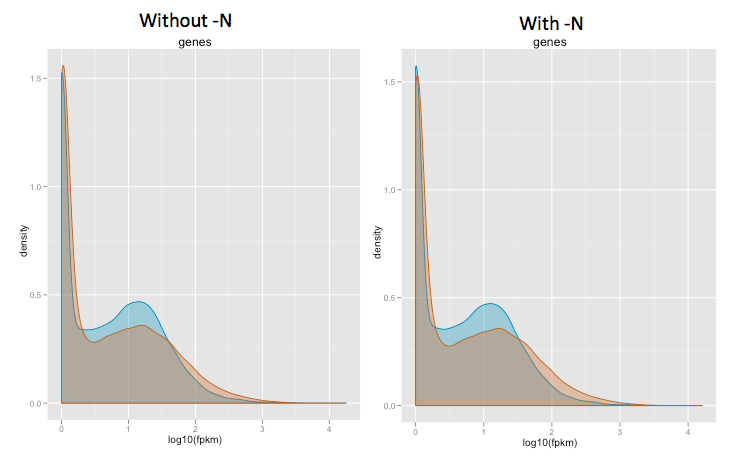
3. Trying to troubleshoot this, I compare cummeRbund density plots with and without "-N", the density plots doesn't change much at all except at very low log10(RPKM) (See picture) But I thought quantile normalization tries to force the two distributions to be very similar?
Any comments/helps will be appreciated!!
I am following the recently published Nature Protocol guideline for analyzing my RNA-seq runs using tophat-cufflink-cuffmerge-cuffdiff (cufflink.1.3.0). I wanted see whether upper quantile normalization improved my results so I also used the "-N" option for Cuffdiff. But Here are some summaries from the "gene_exp.diff" output files with or without the "-N" option:
Condition #ok.status Correlation #Significant DE.Sample.1>Sample.2 DE.Sample.2>sample.1
Without -N 11884 0.6156752 3865 1851 2014
With -N 11885 0.6139065 4435 1620 2815
My questions are:
1. Is it common to see such HUGE variations on the number of significantly DE genes, the numbers of DE sample 1 > sample 2 and sample 2 > sample 1 just for adding this "-N" option?? and since there's such a huge change, which DE gene list should I trust??
2. I noticed there is also "-N" option for cufflink. So at which step should I use such option, and will using "--total-hits-norm" also advisable?
3. Trying to troubleshoot this, I compare cummeRbund density plots with and without "-N", the density plots doesn't change much at all except at very low log10(RPKM) (See picture) But I thought quantile normalization tries to force the two distributions to be very similar?
Any comments/helps will be appreciated!!
Comment