 Tweet
Tweet
Strandness in GTF/GFF file: Strand of the gene, the coding strand of the RNA, the non-template strand
Strandness in column 2 of SAM file: The strand the read being mapped onto the reference
Strandness of Tophat XS:A tag: Strand of the RNA that produce the read.
What I don't understand is that "Strand of the RNA" and "Strand of the gene in GTF". I think they are the same thing?
If I am correct, then for a gene A sit on the +ve strand. Then I should only say the read comes from this gene A if Tophat tag this pair with XS:A:+ve.
Strandness in column 2 of SAM file: The strand the read being mapped onto the reference
Strandness of Tophat XS:A tag: Strand of the RNA that produce the read.
What I don't understand is that "Strand of the RNA" and "Strand of the gene in GTF". I think they are the same thing?
If I am correct, then for a gene A sit on the +ve strand. Then I should only say the read comes from this gene A if Tophat tag this pair with XS:A:+ve.
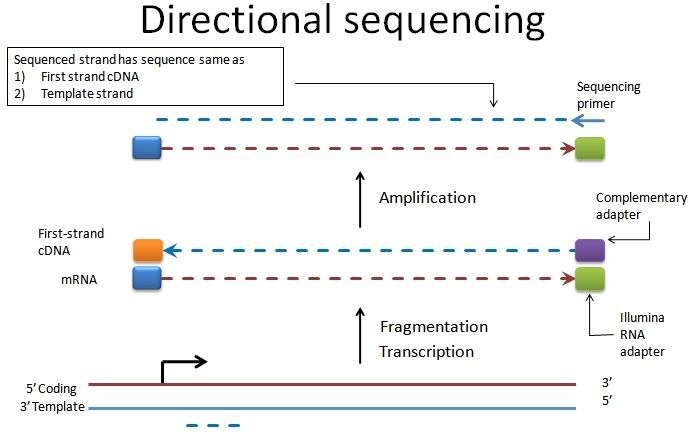
So the read obtained from directional sequencing corresponds to the template strand of the gene (not coding strand). Having said that, I infer the following. However, if we use XS tag in TopHat, we'd get the sense/antisense wrong (thanks Marcowanger for pointing that this was not an HTSeq issue):
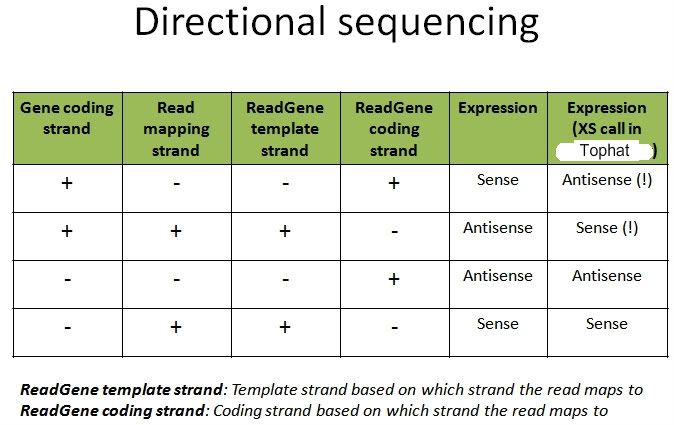
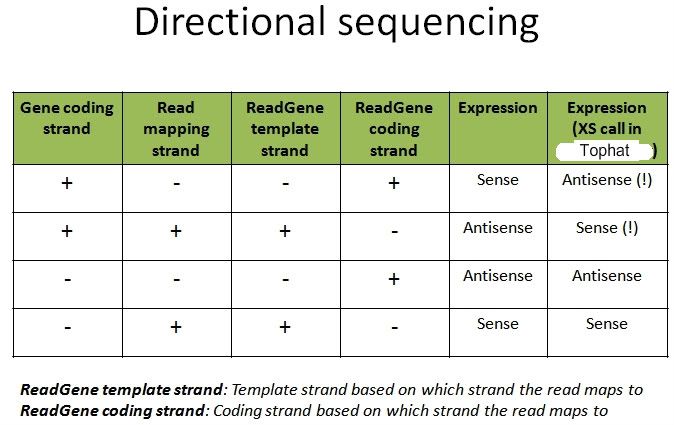
Is my understanding correct? Or am I being mistaken?
Flobpf
UPDATE June 10, 2011: I contacted Illumina and they were confused too. However, finally they (and people at TopHat and our sequencing center) resolved the issue. The reads that come out of the machine have the same sequence as the CODING strand of the DNA and not the template strand.


Comment