 Tweet
Tweet
I'm running ICE and Quiver using tofu_wrap.py, I tried many ways but still failed. I don't understand what error log said and I have attached two log files. If anybody here has any advice I'd be really appreciated.
Thanks,
Ling
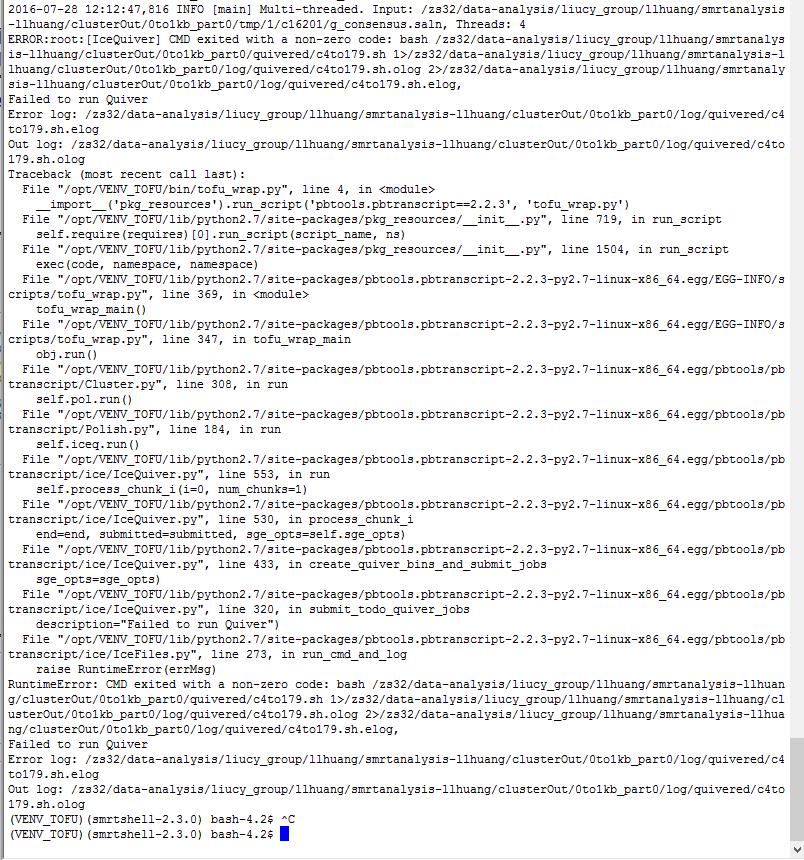
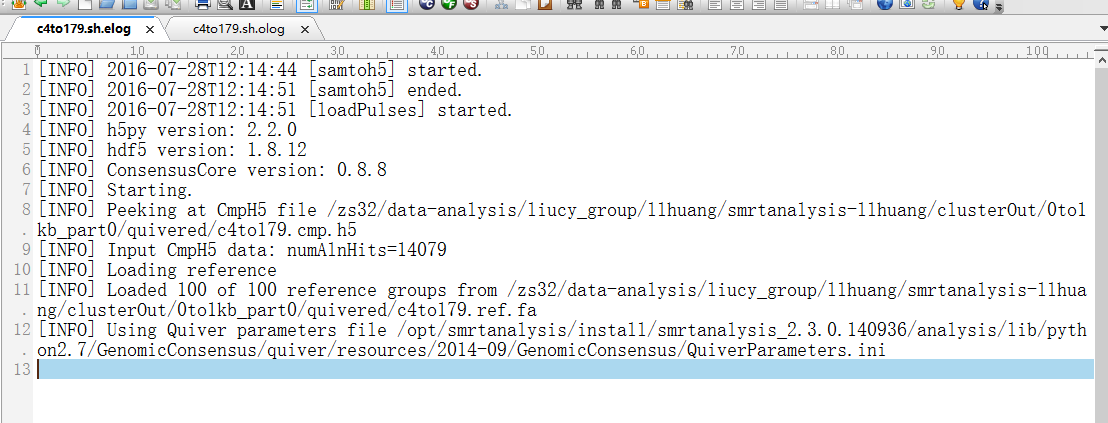
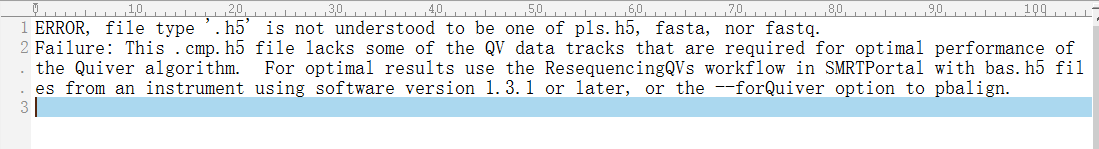
Thanks,
Ling
 I can't understand the cause of the error displayed on screen, as usual, I attach them at below. As you can see, the code has been running for half.Could you explain why this is the case? Thank you!
I can't understand the cause of the error displayed on screen, as usual, I attach them at below. As you can see, the code has been running for half.Could you explain why this is the case? Thank you!
 Hi, bowhan. Just to let you know I finally got the right result. Thank u for helping me all the time.
Hi, bowhan. Just to let you know I finally got the right result. Thank u for helping me all the time.
Comment