 Tweet
Tweet
Hi All,
We have 100bp PE RNAseq data generated on a HiSeq2000 from a library prepped with the Nugen Ovation v2 kit. The samples are from human and rat, with the human samples of lesser quality (RIN<7) whilst the rat are fine. Reads were aligned with bowtie using default options.
Using RSeQC we see the following profiles of counts across transcripts:
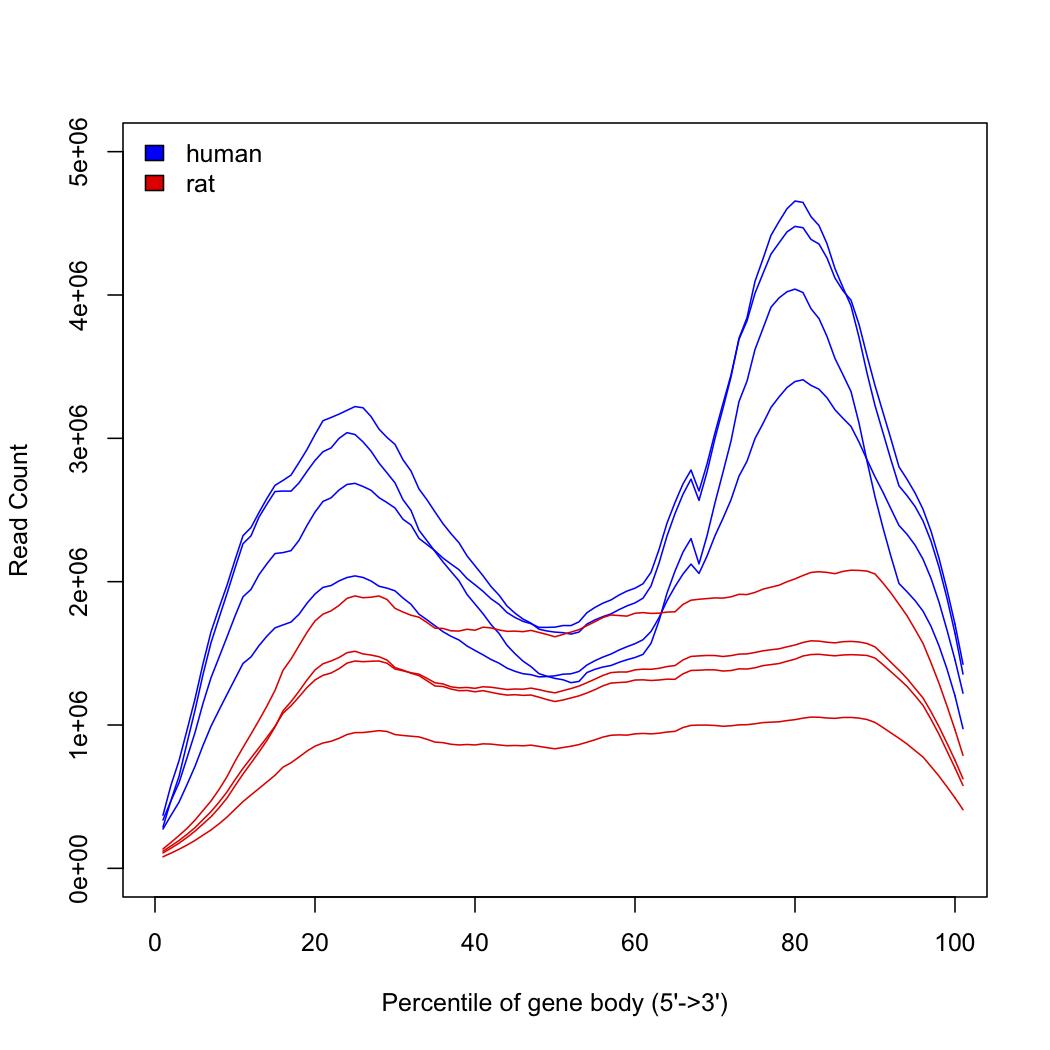
Does anyone have any ideas on how to interpret this? I was expecting a possible 3' bias from the degraded(?) samples and/or the library prep, but wasn't expecting to see both a 3' and 5' bias...
What would you do on the back of this, anything?
Thanks.
We have 100bp PE RNAseq data generated on a HiSeq2000 from a library prepped with the Nugen Ovation v2 kit. The samples are from human and rat, with the human samples of lesser quality (RIN<7) whilst the rat are fine. Reads were aligned with bowtie using default options.
Using RSeQC we see the following profiles of counts across transcripts:
Does anyone have any ideas on how to interpret this? I was expecting a possible 3' bias from the degraded(?) samples and/or the library prep, but wasn't expecting to see both a 3' and 5' bias...
What would you do on the back of this, anything?
Thanks.
Comment