
 Tweet
Tweet
Hi All,
Two questions about RNAseq coverage:
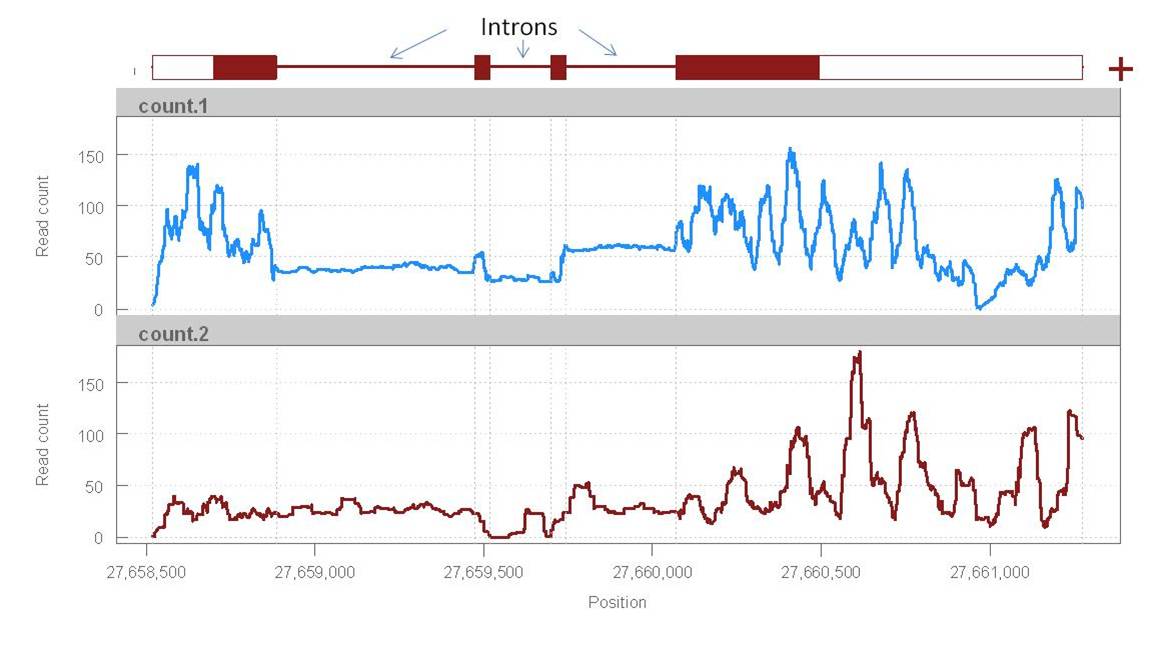
1) Introns: I find that introns get quite a lot of reads mapping to them compared to the exons. Is this expected or is it a sign of something that went wrong? (I would expect introns to be almost invisible).
2) Read distribution: The read count varies a lot in the exons while it seems quite uniform in the introns. Is there any explanation for this?
I attach here a plot of read count vs position (mpileup) in two samples for a gene as an example of what I mean (at a glance, other genes show similar patterns).
Reads & alignments: c.ca 30 million reads, 35 bp pair-end aligned with Tophat.
I realize these issues must have been discussed a number of times but I couldn't find any threads/papers.
Any and all comments welcome!
Dario
Two questions about RNAseq coverage:
1) Introns: I find that introns get quite a lot of reads mapping to them compared to the exons. Is this expected or is it a sign of something that went wrong? (I would expect introns to be almost invisible).
2) Read distribution: The read count varies a lot in the exons while it seems quite uniform in the introns. Is there any explanation for this?
I attach here a plot of read count vs position (mpileup) in two samples for a gene as an example of what I mean (at a glance, other genes show similar patterns).
Reads & alignments: c.ca 30 million reads, 35 bp pair-end aligned with Tophat.
I realize these issues must have been discussed a number of times but I couldn't find any threads/papers.
Any and all comments welcome!
Dario









 . You will always have some amount of contaminating genomic DNA and unprocessed RNA (hnRNA). There are molecular strategies for reducing these (e.g. DNAseI treatment, cytoplasmic RNA isolation, polyA+ selection, etc.) but there will always be some. So, you expect signal across the genome in an RNA-seq library for purely molecular reasons and analytical sources add on top of this (e.g. multi-mapping reads). You can actually see hints of the difference between intergenic noise and intronic noise by examining the data. Intronic noise is expected to be correlated with gene expression level (more expression results in more unprocessed RNA when you take a snapshot of the transcriptome). This is exactly what we see in our RNA-seq libraries. This complicates the interpretation of whether an intron is really being retained as part of an alternative isoform. As others have mentioned, local context is important when interpreting RNA-seq data.
. You will always have some amount of contaminating genomic DNA and unprocessed RNA (hnRNA). There are molecular strategies for reducing these (e.g. DNAseI treatment, cytoplasmic RNA isolation, polyA+ selection, etc.) but there will always be some. So, you expect signal across the genome in an RNA-seq library for purely molecular reasons and analytical sources add on top of this (e.g. multi-mapping reads). You can actually see hints of the difference between intergenic noise and intronic noise by examining the data. Intronic noise is expected to be correlated with gene expression level (more expression results in more unprocessed RNA when you take a snapshot of the transcriptome). This is exactly what we see in our RNA-seq libraries. This complicates the interpretation of whether an intron is really being retained as part of an alternative isoform. As others have mentioned, local context is important when interpreting RNA-seq data.


Comment