
I'd like to introduce a new read-pairing tool, BBMerge, and present a comparison with some similar tools, FLASH, PEAR, COPE, and fastq-join. BBMerge has actually existed since Aug. 2012 but I only recently got around to benchmarking it.
First, what does BBMerge do? Illumina paired reads can be intentionally generated with an insert size (length of template molecule) that is shorter than the sum of the lengths of read 1 and read 2. For example, at JGI, the bulk of our data is 2x150bp fragment libraries designed to have an average insert size of 270bp. Because they overlap, the two reads to be merged into a single, longer read. Single reads are easier to assemble, and longer reads give a better assembly; furthermore, the merging process can detect or correct errors where the reads overlap, yielding a lower error rate than the raw reads. But if the reads are merged incorrectly – yielding a single read with a different length than the actual insert size of the original molecule – errors will be added. So for this process to be useful, it is crucial that reads be merged conservatively enough that the advantages outweigh the disadvantages from incorrect merges.
On to the comparison. I generated 4 million synthetic pairs of 2x150bp reads from E.coli, with errors mimicking the Illumina quality profile. The insert size mean was 270bp and the standard deviation was 30bp, with the min and max insert sizes capped at 3 standard deviations. The exact command was:
randomreads.sh ref=ecoli_K12.fa reads=4000000 paired interleaved out=reads.fq.gz zl=6 len=150 mininsert=180 maxinsert=360 bell maxq=38 midq=28 minq=18 qvariance=16 ow nrate=0.05 maxns=2 maxnlen=1
Then I renamed these reads with names indicating their insert size, e.g. “insert=191” for a pair with insert size 191bp:
bbrename.sh in=reads.fq.gz int renamebyinsert out1=r1.fq out2=r2.fq
Next I merged the reads with each program, varying the stringency along one parameter.
The FLASH command varied the –x parameter from 0.01 (the min allowed) through 0.25 (the default):
/path/FLASH -M 150 -x 0.25 r1.fq r2.fq 1>FLASH.log
The BBMerge command used 5 different flags in terms of decreasing stringency, “strict”, “fast”, default, “loose”, “vloose”:
java -da -Xmx400m -cp /path/ jgi.BBMerge in1=r1.fq in2=r2.fq out=x.fq minoi=150
The PEAR command varied the p-value from 0.0001 to 1 (note that only 5 values were allowed by the program):
pear -n 150 -p 1 -f r1.fq -r r2.fq -o outpear
The COPE command varied the -c (minimum match ratio parameter) from 1.0 (no mismatches) to 0.75 (the default):
./cope -a /dev/shm/r1.fq -b /dev/shm/r2.fq -o /dev/shm/cmerged.fq -2 /dev/shm/r1_unconnected.fq -3 /dev/shm/r2_unconnected.fq -m 0 -s 33 -c 1.0 -u 150 -N 0 1>cope.o 2>&1
The fastq-join (from ea-utils) command line swept -p from 0 to 25 and -m from 4 to 12 parameters; only the -p sweep is shown, as it gave better results. Sample command:
fastq-join r1.fq r2.fq -o fqj -p 0
All programs (except fastq-merge, which does not support the option) were capped at not allowing insert sizes shorter than 150bp. This is because FLASH (which appears to be currently the most popular merger) does not seem to be capable of handling reads with insert sizes shorter than read length. If FLASH is forced to allow such overlaps (using the –O flag) then the results it produces from such reads are 100% wrong, resulting in performance that seems bad to the point of being hard to believe, which would undermine this analysis, so I limited the range of insert sizes to at minimum 180, which FLASH can handle. The output was graded by my grading script to determine the percent correct and incorrect:
grademerge.sh in=x.fq
Then the results were plotted in a ROC curve, attached. The X-axis is logarithmic in one figure and linear in the other.
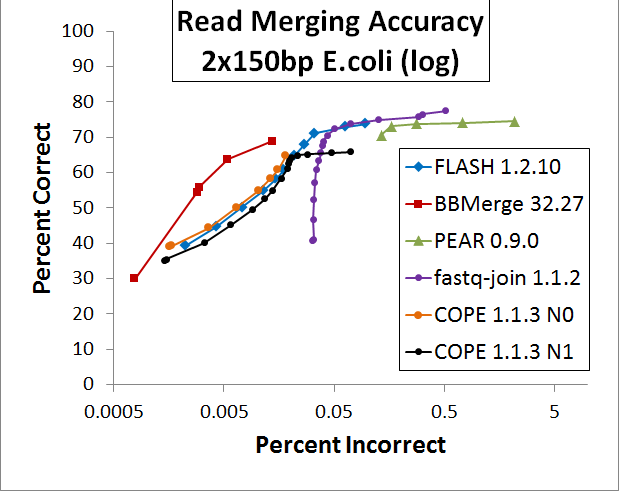

FLASH achieves a slightly high merging rate at its default sensitivity, but does so at the expense of an extremely high false positive rate. BBMerge has a systematically lower false positive rate than FLASH at any sensitivity. fastq-join yields the highest merging rate, at the cost of the highest false-positive rate, second only to PEAR. COPE is very competitive with FLASH and strictly exceeds it with "N=0"; unfortunately it is unstable at that setting. COPE is stable at "N=1" (the default) but is generally inferior to FLASH at that setting. PEAR appears to be inferior to all other options across the board.
Next, I tested the speed. Since BBMerge, FLASH, and PEAR are multithreaded, I tested with various numbers of threads to determine peak performance. The tests were run on a 4x8-core Sandy Bridge E node with no other processes running; these had hyperthreading enabled, for 32 cores and 64 threads. Input came from a ramdisk and was sent to a ramdisk, so I/O bandwidth was not limiting. The results attached to this post indicate what happens when input was from local disk and output went to a networked filesystem, but that was I/O limited; the image embedded below reflects the ramdisk run.
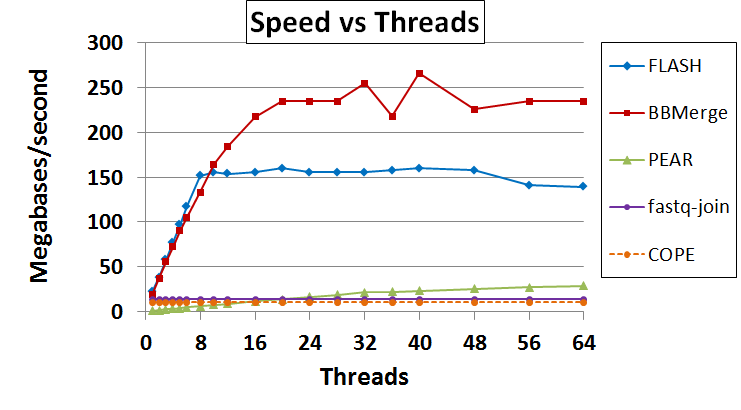
FLASH has a slight advantage with a low number of threads, but BBMerge ultimately scales better to reach a higher peak of 235 megabasepairs/s (Mbp/s) versus FLASH’s 160 Mbp/s. BBMerge’s peak came at 20 threads and FLASH’s at 36 threads. 235 Mbp/s in this case equates to 499 megabytes per second and BBMerge was probably input-limited (by CPU usage of the input thread) at that point. PEAR was very slow so I did not run the full suite of speed testing on it, but it achieved 11.8 Mbp/s at 16 threads (on slightly different hardware). The single-threaded programs (COPE and fastq-join) beat PEAR up to around 16 threads, but never touched BBMerge or FLASH. Interestingly, PEAR is the only program that scales well with hyperthreading; I speculate that the program does a lot of floating-point operations.
In conclusion, for this dataset, BBMerge had substantially better specificity at similar sensitivity to all competitors. fastq-join had the highest merge rate, by a slight margin, but at the cost of a false positive rate that I would consider unusable in a production environment. The speeds of BBMerge and FLASH are similar per thread, with FLASH being around 10% higher; but BBMerge appears to be more scalable, and as such exceeded 150% of FLASH's peak throughput. So, for this dataset, unless false positives are irrelevant, BBMerge wins.
It appears imprudent to use FLASH (or most other mergers) with the default sensitivity, due to the huge increase in false positives for virtually no gain in true positives. On the ROC curve, FLASH's rightmost point is the default “-x 0.25”, while the third from the right is “-x 0.15”, which gives a much better signal-to-noise ratio of 33.3dB and 71.1% merged compared to the default 28.8dB and 73.8% merged. If you desire the highest merge rate regardless of false-positives, fastq-join appears optimal. For libraries that have all N-containing reads removed, it may be that COPE is slightly better than FLASH, though I have not verified this. If false-positives are detrimental to your project, I recommend BBMerge on default sensitivity, which gave a 42.7dB SNR and 55.8% merge rate for this data. The merge rate can be increased using the "loose" or "vloose" flags, at the expense of more false-positives, but I believe that for most projects, minimizing noise is more important than maximizing signal.
BBMerge is available with the BBTools package here:
On Linux systems you can simply run the shellscript, like this:
bbmerge.sh in1=read1.fq in2=read2.fq out=merged.fq
BBMerge supports interleaved reads, ASCII-33 or 64, gzip, fasta, scarf, and various other file formats. It also makes handy insert-size distribution histograms. Everything in the BBTools package is compatible with any OS that supports Java - Linux, Windows, Mac, even cell phones. The shellscripts may only work in Linux, but the actual programs will run anywhere. For more details, run the shellscript with no arguments, or post a question here.
EDIT: Results updated March 25, 2015, on a poster:
First, what does BBMerge do? Illumina paired reads can be intentionally generated with an insert size (length of template molecule) that is shorter than the sum of the lengths of read 1 and read 2. For example, at JGI, the bulk of our data is 2x150bp fragment libraries designed to have an average insert size of 270bp. Because they overlap, the two reads to be merged into a single, longer read. Single reads are easier to assemble, and longer reads give a better assembly; furthermore, the merging process can detect or correct errors where the reads overlap, yielding a lower error rate than the raw reads. But if the reads are merged incorrectly – yielding a single read with a different length than the actual insert size of the original molecule – errors will be added. So for this process to be useful, it is crucial that reads be merged conservatively enough that the advantages outweigh the disadvantages from incorrect merges.
On to the comparison. I generated 4 million synthetic pairs of 2x150bp reads from E.coli, with errors mimicking the Illumina quality profile. The insert size mean was 270bp and the standard deviation was 30bp, with the min and max insert sizes capped at 3 standard deviations. The exact command was:
randomreads.sh ref=ecoli_K12.fa reads=4000000 paired interleaved out=reads.fq.gz zl=6 len=150 mininsert=180 maxinsert=360 bell maxq=38 midq=28 minq=18 qvariance=16 ow nrate=0.05 maxns=2 maxnlen=1
Then I renamed these reads with names indicating their insert size, e.g. “insert=191” for a pair with insert size 191bp:
bbrename.sh in=reads.fq.gz int renamebyinsert out1=r1.fq out2=r2.fq
Next I merged the reads with each program, varying the stringency along one parameter.
The FLASH command varied the –x parameter from 0.01 (the min allowed) through 0.25 (the default):
/path/FLASH -M 150 -x 0.25 r1.fq r2.fq 1>FLASH.log
The BBMerge command used 5 different flags in terms of decreasing stringency, “strict”, “fast”, default, “loose”, “vloose”:
java -da -Xmx400m -cp /path/ jgi.BBMerge in1=r1.fq in2=r2.fq out=x.fq minoi=150
The PEAR command varied the p-value from 0.0001 to 1 (note that only 5 values were allowed by the program):
pear -n 150 -p 1 -f r1.fq -r r2.fq -o outpear
The COPE command varied the -c (minimum match ratio parameter) from 1.0 (no mismatches) to 0.75 (the default):
./cope -a /dev/shm/r1.fq -b /dev/shm/r2.fq -o /dev/shm/cmerged.fq -2 /dev/shm/r1_unconnected.fq -3 /dev/shm/r2_unconnected.fq -m 0 -s 33 -c 1.0 -u 150 -N 0 1>cope.o 2>&1
The fastq-join (from ea-utils) command line swept -p from 0 to 25 and -m from 4 to 12 parameters; only the -p sweep is shown, as it gave better results. Sample command:
fastq-join r1.fq r2.fq -o fqj -p 0
All programs (except fastq-merge, which does not support the option) were capped at not allowing insert sizes shorter than 150bp. This is because FLASH (which appears to be currently the most popular merger) does not seem to be capable of handling reads with insert sizes shorter than read length. If FLASH is forced to allow such overlaps (using the –O flag) then the results it produces from such reads are 100% wrong, resulting in performance that seems bad to the point of being hard to believe, which would undermine this analysis, so I limited the range of insert sizes to at minimum 180, which FLASH can handle. The output was graded by my grading script to determine the percent correct and incorrect:
grademerge.sh in=x.fq
Then the results were plotted in a ROC curve, attached. The X-axis is logarithmic in one figure and linear in the other.
FLASH achieves a slightly high merging rate at its default sensitivity, but does so at the expense of an extremely high false positive rate. BBMerge has a systematically lower false positive rate than FLASH at any sensitivity. fastq-join yields the highest merging rate, at the cost of the highest false-positive rate, second only to PEAR. COPE is very competitive with FLASH and strictly exceeds it with "N=0"; unfortunately it is unstable at that setting. COPE is stable at "N=1" (the default) but is generally inferior to FLASH at that setting. PEAR appears to be inferior to all other options across the board.
Next, I tested the speed. Since BBMerge, FLASH, and PEAR are multithreaded, I tested with various numbers of threads to determine peak performance. The tests were run on a 4x8-core Sandy Bridge E node with no other processes running; these had hyperthreading enabled, for 32 cores and 64 threads. Input came from a ramdisk and was sent to a ramdisk, so I/O bandwidth was not limiting. The results attached to this post indicate what happens when input was from local disk and output went to a networked filesystem, but that was I/O limited; the image embedded below reflects the ramdisk run.
FLASH has a slight advantage with a low number of threads, but BBMerge ultimately scales better to reach a higher peak of 235 megabasepairs/s (Mbp/s) versus FLASH’s 160 Mbp/s. BBMerge’s peak came at 20 threads and FLASH’s at 36 threads. 235 Mbp/s in this case equates to 499 megabytes per second and BBMerge was probably input-limited (by CPU usage of the input thread) at that point. PEAR was very slow so I did not run the full suite of speed testing on it, but it achieved 11.8 Mbp/s at 16 threads (on slightly different hardware). The single-threaded programs (COPE and fastq-join) beat PEAR up to around 16 threads, but never touched BBMerge or FLASH. Interestingly, PEAR is the only program that scales well with hyperthreading; I speculate that the program does a lot of floating-point operations.
In conclusion, for this dataset, BBMerge had substantially better specificity at similar sensitivity to all competitors. fastq-join had the highest merge rate, by a slight margin, but at the cost of a false positive rate that I would consider unusable in a production environment. The speeds of BBMerge and FLASH are similar per thread, with FLASH being around 10% higher; but BBMerge appears to be more scalable, and as such exceeded 150% of FLASH's peak throughput. So, for this dataset, unless false positives are irrelevant, BBMerge wins.
It appears imprudent to use FLASH (or most other mergers) with the default sensitivity, due to the huge increase in false positives for virtually no gain in true positives. On the ROC curve, FLASH's rightmost point is the default “-x 0.25”, while the third from the right is “-x 0.15”, which gives a much better signal-to-noise ratio of 33.3dB and 71.1% merged compared to the default 28.8dB and 73.8% merged. If you desire the highest merge rate regardless of false-positives, fastq-join appears optimal. For libraries that have all N-containing reads removed, it may be that COPE is slightly better than FLASH, though I have not verified this. If false-positives are detrimental to your project, I recommend BBMerge on default sensitivity, which gave a 42.7dB SNR and 55.8% merge rate for this data. The merge rate can be increased using the "loose" or "vloose" flags, at the expense of more false-positives, but I believe that for most projects, minimizing noise is more important than maximizing signal.
BBMerge is available with the BBTools package here:

On Linux systems you can simply run the shellscript, like this:
bbmerge.sh in1=read1.fq in2=read2.fq out=merged.fq
BBMerge supports interleaved reads, ASCII-33 or 64, gzip, fasta, scarf, and various other file formats. It also makes handy insert-size distribution histograms. Everything in the BBTools package is compatible with any OS that supports Java - Linux, Windows, Mac, even cell phones. The shellscripts may only work in Linux, but the actual programs will run anywhere. For more details, run the shellscript with no arguments, or post a question here.
EDIT: Results updated March 25, 2015, on a poster:






Comment