
CalcTrueQuality is a new member of the BBTools package. In light of the quality-score issues with the NextSeq platform, and the possibility of future Illumina platforms (HiSeq 3000 and 4000) also using quantized quality scores, I developed it for recalibrating the scores to ensure accuracy and restore the full range of values.
The usage is fairly simple. I will walk through how I recalibrated the reads used in the attached images. First adapter-trim the reads:
bbduk.sh -Xmx1g in=reads.fq out=trimmed.fq ref=adapters.fa tbo tpe k=23 mink=11 hdist=1 ftm=5
This is very important because adapter sequence will appear to be errors during mapping, so it will mess up the calibration statistics. Do not quality-trim or quality filter. The "ftm=5" flag is optional; it will trim the last base off of 151bp reads; that base is very low quality. Specifically, ftm=5 will trim reads so that their length is equal to zero modulo 5, and ignore reads that are already 100bp or 150bp, etc. Next, map the reads to a reference:
bbmap.sh in=trimmed.fq outm=mapped.sam ref=ref.fa ignorequality maxindel=100 minratio=0.4 ambig=toss qahist=qahist_raw.txt qhist=qhist_raw.txt mhist=mhist_raw.txt
A finished reference is optimal, but not required; any assembly will be sufficient. For paired reads, I recommend using the "po" flag to only include proper pairs in the output. If you have a lot of input you can just map a fraction of it, with e.g. the flag "reads=20m" to only use the first 20 million reads (or pairs). You don't have to use BBMap for this, but you do need to use an aligner that produces cigar strings with "=" and "X" symbols for match and mismatch, not just "M" for everything.
Then, generate recalibration matrices:
calctruequality.sh in=mapped.sam
This will analyze the sam file and write some small files to /ref/qual/ (you can change the location to write to with the "path" flag). You can also give the program multiple sam files with a comma-delimited list; the more data, the more precise the calibration.
Lastly, recalibrate the reads:
bbduk.sh in=trimmed.fq out=recalibrated.fq recalibrate
The reads that you map to generate the calibration matrices do not need to be the same as the reads you are recalibrating. For example, if you multiplexed 10 samples in a lane, you could just map one of them and recalibrate the others based on it. But certainly it's best to do the mapping with reads from the same run, and ideally, the actual reads you are recalibrating.
This is what the NextSeq V1 data looked before recalibration (but after adapter-trimming):
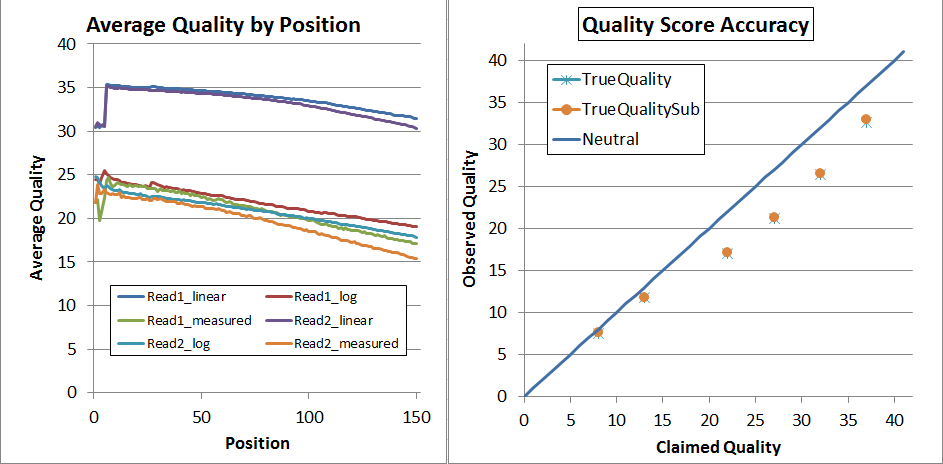
The left is "qhist" and the right is "qahist", both outputs from BBMap. For qhist, the "log" lines are based on translating the quality score to probability, then taking the average, then translating back to a quality score, so perfectly accurate quality scores will have for example the "Read1_log" line exactly overlaying the "Read1_measured" line.
With perfectly accurate quality scores, the yellow points on the qahist graph will overlay the dark blue line. Points under the blue line indicate inflated quality scores. For example, the upper-right yellow point is calculated by counting the number of times bases with Q37 map with a match or a mismatch. TrueQualitySub is based only on mismatches, while TrueQuality also counts indels as errors.
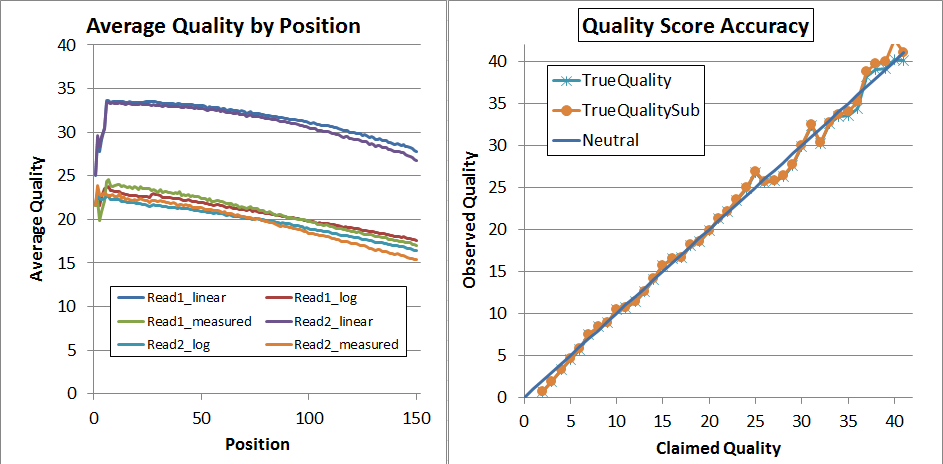
After recalibration, the data looks much better, and has expanded to the full range of 2 through 41:
Finally, since that's not quite perfect, I decided to try a second pass of recalibration. I don't think that's generally necessary and it requires you to regenerate the calibration matrices (you can't use the same ones again!) but it did lead to very nice results:
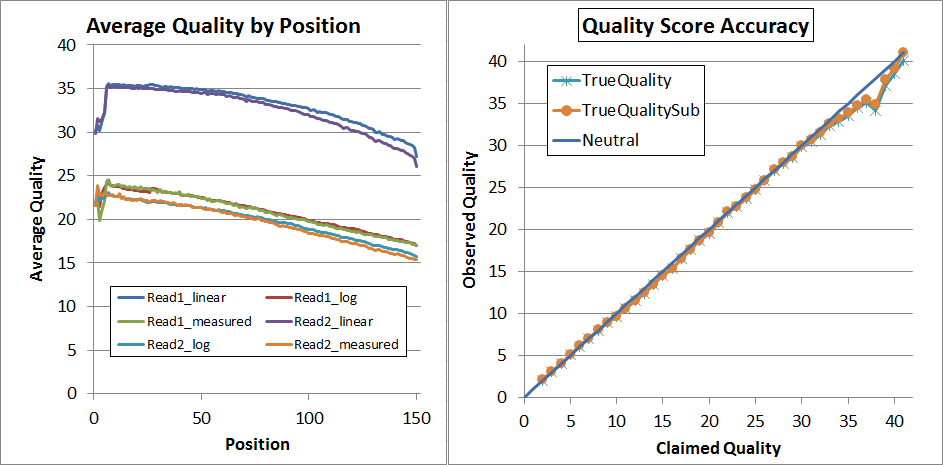
As you can see, up to Q34 the quality scores are pretty much dead-on in the qahist, and in qhist, the "log" lines are now very close to the "measured" lines.
This program will work with any Illumina reads, not just NextSeq. Right now I don't recommend it for platforms that exhibit indel-type errors because those are not used in the recalibration.
The usage is fairly simple. I will walk through how I recalibrated the reads used in the attached images. First adapter-trim the reads:
bbduk.sh -Xmx1g in=reads.fq out=trimmed.fq ref=adapters.fa tbo tpe k=23 mink=11 hdist=1 ftm=5
This is very important because adapter sequence will appear to be errors during mapping, so it will mess up the calibration statistics. Do not quality-trim or quality filter. The "ftm=5" flag is optional; it will trim the last base off of 151bp reads; that base is very low quality. Specifically, ftm=5 will trim reads so that their length is equal to zero modulo 5, and ignore reads that are already 100bp or 150bp, etc. Next, map the reads to a reference:
bbmap.sh in=trimmed.fq outm=mapped.sam ref=ref.fa ignorequality maxindel=100 minratio=0.4 ambig=toss qahist=qahist_raw.txt qhist=qhist_raw.txt mhist=mhist_raw.txt
A finished reference is optimal, but not required; any assembly will be sufficient. For paired reads, I recommend using the "po" flag to only include proper pairs in the output. If you have a lot of input you can just map a fraction of it, with e.g. the flag "reads=20m" to only use the first 20 million reads (or pairs). You don't have to use BBMap for this, but you do need to use an aligner that produces cigar strings with "=" and "X" symbols for match and mismatch, not just "M" for everything.
Then, generate recalibration matrices:
calctruequality.sh in=mapped.sam
This will analyze the sam file and write some small files to /ref/qual/ (you can change the location to write to with the "path" flag). You can also give the program multiple sam files with a comma-delimited list; the more data, the more precise the calibration.
Lastly, recalibrate the reads:
bbduk.sh in=trimmed.fq out=recalibrated.fq recalibrate
The reads that you map to generate the calibration matrices do not need to be the same as the reads you are recalibrating. For example, if you multiplexed 10 samples in a lane, you could just map one of them and recalibrate the others based on it. But certainly it's best to do the mapping with reads from the same run, and ideally, the actual reads you are recalibrating.
This is what the NextSeq V1 data looked before recalibration (but after adapter-trimming):
The left is "qhist" and the right is "qahist", both outputs from BBMap. For qhist, the "log" lines are based on translating the quality score to probability, then taking the average, then translating back to a quality score, so perfectly accurate quality scores will have for example the "Read1_log" line exactly overlaying the "Read1_measured" line.
With perfectly accurate quality scores, the yellow points on the qahist graph will overlay the dark blue line. Points under the blue line indicate inflated quality scores. For example, the upper-right yellow point is calculated by counting the number of times bases with Q37 map with a match or a mismatch. TrueQualitySub is based only on mismatches, while TrueQuality also counts indels as errors.
After recalibration, the data looks much better, and has expanded to the full range of 2 through 41:
Finally, since that's not quite perfect, I decided to try a second pass of recalibration. I don't think that's generally necessary and it requires you to regenerate the calibration matrices (you can't use the same ones again!) but it did lead to very nice results:
As you can see, up to Q34 the quality scores are pretty much dead-on in the qahist, and in qhist, the "log" lines are now very close to the "measured" lines.
This program will work with any Illumina reads, not just NextSeq. Right now I don't recommend it for platforms that exhibit indel-type errors because those are not used in the recalibration.




Comment