
 Tweet
Tweet
I'm running amplicons (v4 16s, dual index, custom sequencing primers, Kozich/Schloss primers). I'd been having good luck so was dialing up my clustering density and dialing down my phiX. And then everything fell apart. I have one project that has very low diversity (microbial community wise) that I don't want to run by itself. So my last successful run and this current run that keep failing are both made up of 25% low diversity project samples and 75% regular diversity samples (which are still low diversity as far as sequencing goes).
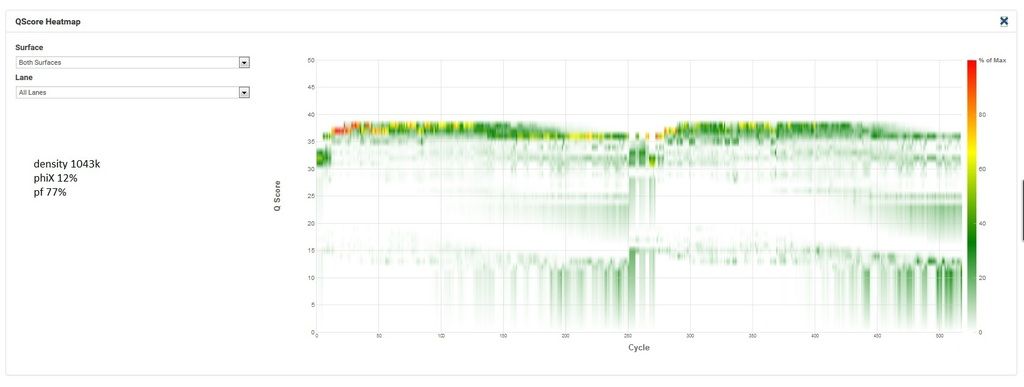
Here's the q30 heatmap for the successful run:
Now the unsuccessful. This is the second attempt at this exact pool, the first had similar cluster density and phiX as the successful run but R2 was worthless (lots of Ns and 0% phiX aligning in R2) so my FAS wanted me to try rerunning it with lower density and much more phiX.
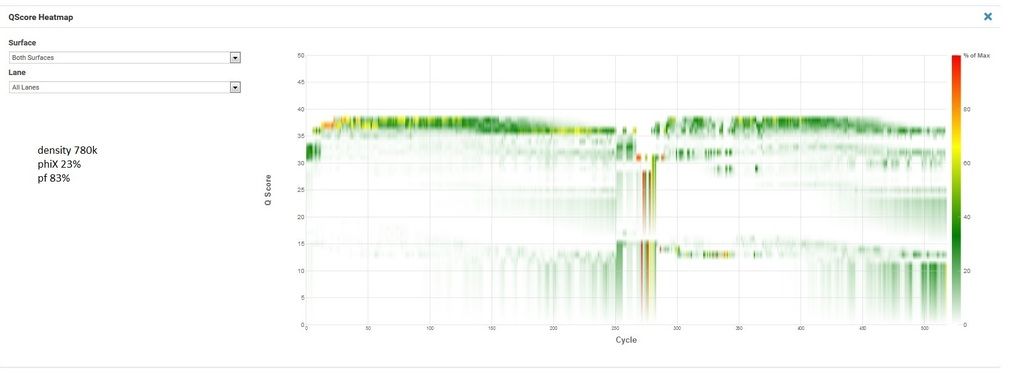
R1, I1, and I2 are all great. The indexing looks good (I'm multiplexing 120 samples and recovering 115 which is well within my expectations). R2 is bad. And no phiX is aligned. The lack of phiX and the indexing make me pretty confident it's not my library. But I'm at a loss what else it could be. My FAS is kicking this up to second level support, but I'd love to hear if anyone here has had this happen.
Here's the q30 heatmap for the successful run:

Now the unsuccessful. This is the second attempt at this exact pool, the first had similar cluster density and phiX as the successful run but R2 was worthless (lots of Ns and 0% phiX aligning in R2) so my FAS wanted me to try rerunning it with lower density and much more phiX.

R1, I1, and I2 are all great. The indexing looks good (I'm multiplexing 120 samples and recovering 115 which is well within my expectations). R2 is bad. And no phiX is aligned. The lack of phiX and the indexing make me pretty confident it's not my library. But I'm at a loss what else it could be. My FAS is kicking this up to second level support, but I'd love to hear if anyone here has had this happen.







Comment