 Tweet
Tweet
Hello everyone,
my name is Bastian and I have a question concerning the pipeline of sequence quality improvement and control.
My Experiment: RNA SEQ 150 bp paired-end sequencing on Illumina Hiseq4000 platform (genome size: approx. 100 Mbp) 30M reads
My pipeline so far:
I received 'clean data' from my company so they claim adaptors have been removed already.
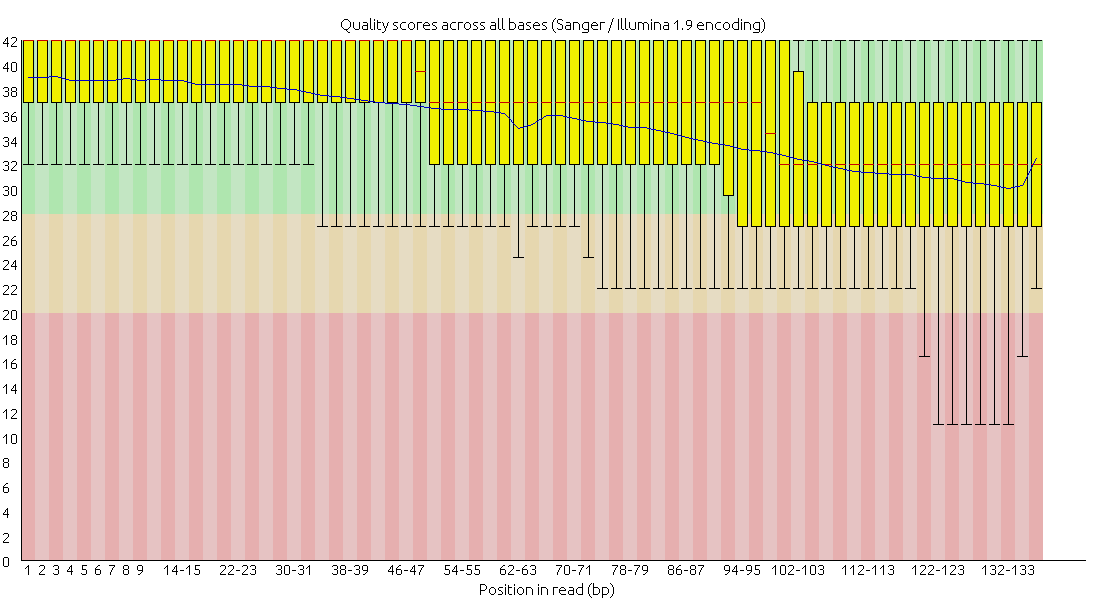
After checking the sequence quality with Fastqc, I used Trimmomatic to further improve sequence quality (HEADCROP:14 SLIDINGWINDOW:4:15 MINLEN:50).
My problem:
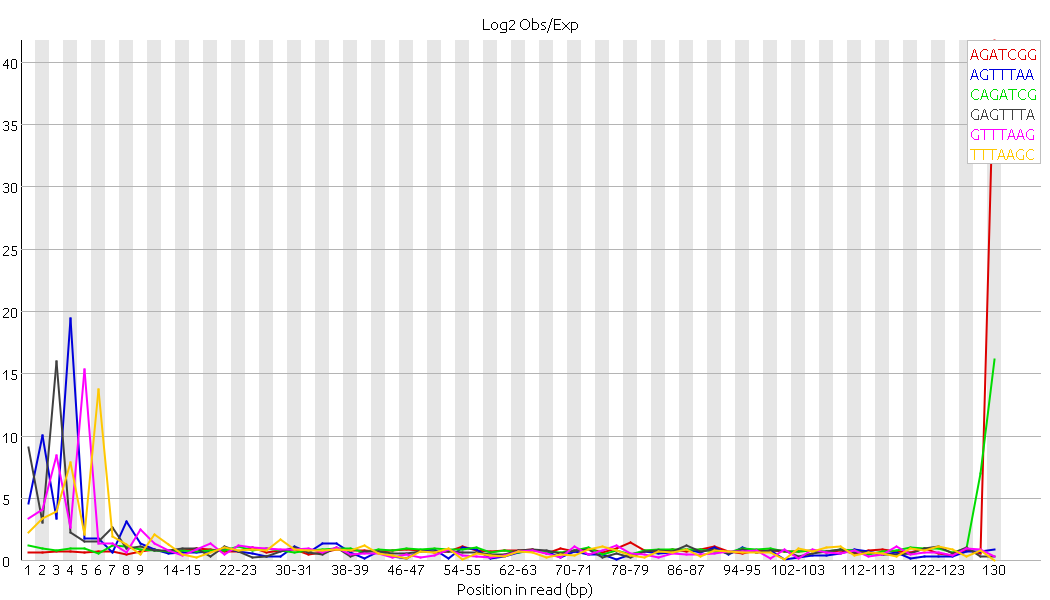
If I check my trimmed sequences with Fastqc I still have an error symbol for Sequence duplication levels and kmer content (especially this peak at 130 bp) --> Fastqc attached
My question(s):
- Are those 'errors' really relevant for assembly? Especially concerning kmer content I found different opinions.
- Which programs would you suggest to get rid of these errors? I tried abundance filtering from khmer and got a little improvement, but still the error warnings are there.
Thank you very much (in advance) and best regards ,
,
Bastian
forward paired: http://www.directupload.net/file/d/4...dld4dd_png.htm
http://www.directupload.net/file/d/4...5d9cxz_png.htm http://www.directupload.net/file/d/4...epa2qu_png.htm
reverse paired: http://www.directupload.net/file/d/4...z7prvg_png.htm
my name is Bastian and I have a question concerning the pipeline of sequence quality improvement and control.
My Experiment: RNA SEQ 150 bp paired-end sequencing on Illumina Hiseq4000 platform (genome size: approx. 100 Mbp) 30M reads
My pipeline so far:
I received 'clean data' from my company so they claim adaptors have been removed already.
After checking the sequence quality with Fastqc, I used Trimmomatic to further improve sequence quality (HEADCROP:14 SLIDINGWINDOW:4:15 MINLEN:50).
My problem:
If I check my trimmed sequences with Fastqc I still have an error symbol for Sequence duplication levels and kmer content (especially this peak at 130 bp) --> Fastqc attached
My question(s):
- Are those 'errors' really relevant for assembly? Especially concerning kmer content I found different opinions.
- Which programs would you suggest to get rid of these errors? I tried abundance filtering from khmer and got a little improvement, but still the error warnings are there.
Thank you very much (in advance) and best regards
,Bastian
forward paired: http://www.directupload.net/file/d/4...dld4dd_png.htm
http://www.directupload.net/file/d/4...5d9cxz_png.htm http://www.directupload.net/file/d/4...epa2qu_png.htm
reverse paired: http://www.directupload.net/file/d/4...z7prvg_png.htm










Comment